


El síndrome anémico en la infancia requiere una orientación diagnóstica y un tratamiento urgente guiado por protocolos sistemáticos, evitando así la realización de pruebas complementarias innecesarias.
Niña de 4 años con astenia, fiebre intermitente y dolor abdominal de 7 días de evolución. Impresiona de regular estado general, presentando ictericia mucocutánea, un soplo III/VI/iv y un abdomen doloroso con hepatoesplenomegalia.
En la analítica se observó una anemia hiporregenerativa con aumento de LDH y bilirrubina indirecta.
El test de Coombs fue negativo, visualizándose esferocitos en el frotis de sangre periférica. Las IgM e IgG fueron positivas para Parvovirus B19 y la IgM para el virus de Epstein-Barr, llegándose al diagnóstico de crisis aplásica en una paciente con esferocitosis hereditaria. No precisó tratamiento específico.
Ante la sospecha de síndrome anémico en urgencias debemos actuar siguiendo la secuencia ABCDE. Gracias a la anamnesis, la exploración y la analítica básica realizaremos una aproximación diagnóstica inicial. Las pruebas etiológicas específicas deben basarse en este primer estudio.
Anemic syndrome in childhood requires a diagnosis and urgent treatment guided by systematic protocols that can avoid unnecessary additional testing.
The case of a 4 year-old girl with fatigue and intermittent fever of 7 days duration, accompanied by abdominal pain is presented. She had regular general health status, with mucocutaneous jaundice, a grade III/VI/iv murmur, and painful abdomen with hepatosplenomegaly.
The blood analysis showed a hypo-regenerative anemia with increased LDH and indirect bilirubin.
The Coombs Test was negative, with spherocytes being observed in the peripheral blood smear. The IgM and IgG were positive for parvovirus B19 IgM and Epstein Barr virus, leading to the diagnosis of aplastic crisis in a patient with hereditary spherocytosis. No specific treatment was required.
Under the suspicion of anemic syndrome in emergencies, the ABCDE sequence must be followed. Through the history, physical examination and basic laboratory tests, an initial diagnostic approach can be made. Specific etiological tests should be based on this first study.
La anemia es una entidad frecuente, que puede presentarse como un hallazgo casual, pudiendo estudiarse de forma diferida o en forma de síndrome anémico, con letargia, palidez, irritabilidad, pobre ingesta oral, taquicardia o datos de hemólisis. En este último caso será necesaria una orientación diagnóstica y un tratamiento urgente, evitando la realización de pruebas diagnósticas innecesarias.
Caso clínicoSe presenta el caso de una niña de 4 años, que acudió a urgencias por fiebre intermitente y astenia de 7 días de evolución, acompañada de dolor abdominal generalizado, que había empeorado en las últimas 24 h. Como antecedente de interés, refería una ictericia neonatal que había precisado fototerapia.
A su llegada, presentaba regular estado general, con un tinte pálido ictérico de piel y mucosas. Se detectó un soplo sistólico iii/vi a la auscultación cardíaca. La paciente refería dolor abdominal generalizado, palpándose polo de bazo e hígado. Presentaba una temperatura de 37,3°C, una presión arterial de 103/70mmHg y una frecuencia cardíaca de 132 lpm. El resto de la exploración resultó normal.
Ante la sospecha de un síndrome anémico en el contexto de una infección en una paciente previamente sana, se realizó un análisis de sangre que incluía hemograma con reticulocitos, frotis de sangre periférica y estudio bioquímico de hemólisis.
En el hemograma destacaba una hemoglobina de 4g/dl, hematocrito 11,6%, VCM 71,4 fl, CHCM 39,2g/dl, leucocitos 20.810/l, neutrófilos 13.670/l, cayados 9% y plaquetas 366.000/μl. El valor de los reticulocitos era del 0,41% y en el frotis de sangre periférica se visualizaron microcitosis, anisocitosis, hipercromía y esferocitos.
En la bioquímica, encontramos urea de 14mg/dl, creatinina 0,5mg/dl, bilirrubina total 1,6mg/dl, bilirrubina conjugada 0,4mg/dl, bilirrubina no conjugada 1,2mg/dl, LDH 481 U/l, transaminasas normales, gammaglutamil transpeptidasa 68 U/l, fósforo 2,4mg/dl, iones normales y ácido úrico 4,7mg/dl, procalcitonina 0,14ng/ml y proteína C reactiva 0,9mg/dl.
Debido al bajo valor de hemoglobina en una paciente con características de shock clínico, se realizaron pruebas cruzadas y soporte transfusional con concentrado de hematíes a 15ml/kg, con mejoría del cuadro clínico.
Ante el hallazgo de una anemia hiporregenerativa, se amplió el estudio con la determinación de hierro, ácido fólico, vitamina B12 y hormonas tiroideas, cuyos resultados fueron normales.
Nuestra paciente presentaba datos de hemólisis, realizándose por tanto un test de Coombs directo, cuyo resultado fue negativo, y se obtuvo el valor de glucosa 6-fosfato deshidrogenasa y el estudio de hemoglobinas, cuyos resultados fueron no patológicos. La haptoglobina estaba disminuida (10mg/dl) y tanto el test de fragilidad osmótica como el test de hemólisis con glucosa/solución salina fueron patológicos.
Tanto la IgM y la IgG para Parvovirus B19, como la IgM para virus de Epstein-Barr virus fueron positivas.
La paciente permaneció ingresada 5 días, sin precisar tratamiento específico, controlándola posteriormente en el servicio de hematología. El diagnóstico final fue crisis aplásica por Parvovirus B19 y virus de Epstein-Barr en una paciente con esferocitosis hereditaria.
DiscusiónAnte la sospecha de un síndrome anémico en urgencias, debemos valorar la necesidad de estabilización inicial con ayuda del triángulo de evaluación pediátrica y la secuencia ABCDE (algoritmo 1, fig. 1)1.

Cuando la anemia se produce de forma aguda (generalmente por hemorragia activa), los pacientes mueren por hipovolemia e hipoperfusión tisular. En ellos, la primera medida será reponer la volemia, no normalizar la hemoglobina. Las indicaciones de transfusión vienen determinadas en la tabla 12-4.
Indicaciones de transfusión
| Indicaciones de transfusión |
| < 4 meses |
| Hemorragia aguda con pérdida de ≥ 10% de la volemia o síntomas de hipoxia que no responde a la administración de fluidos |
| Hb < 7 g/dl con clínica de anemia (apneas, escasa ganancia ponderal, taquicardia, actividad disminuida) |
| Hb < 10 g/dl en: |
| Anemia perioperatoria |
| Shock clínico o disminución severa de la presión arterial |
| 1.ª semana de vida y clínica de anemia |
| Hb < 13 g/dl en pacientes con cardiopatía congénita cianótica grave o ECMO |
| > 4 meses |
| Pérdida sanguínea aguda con síntomas clínicos de hipoxia tras corrección de hipovolemia con cristaloides o coloides |
| Pérdida intraoperatoria ≥ 15% de la volemia |
| Hb < 7 g/dl en pacientes con anemia crónica (congénita o adquirida) que no responde a tratamiento médico o en pacientes en tratamiento con QT o RT |
| Hb < 10 g/dl en: |
| Postoperatorio con signos de anemia |
| Shock clínico o hipotensión grave |
| Preoperatorio en pacientes con transfusiones crónicas |
| Hb < 12 g/dl y enfermedad cardiopulmonar grave |
| Previo a la obtención de precursores hematopoyéticos |
| Determinadas situaciones en anemia drepanocítica |
ECMO: oxigenación por membrana extracorpórea; Hb: hemoglobina; QT: quimioterapia; RT: radioterapia.
Adaptado de: García García1
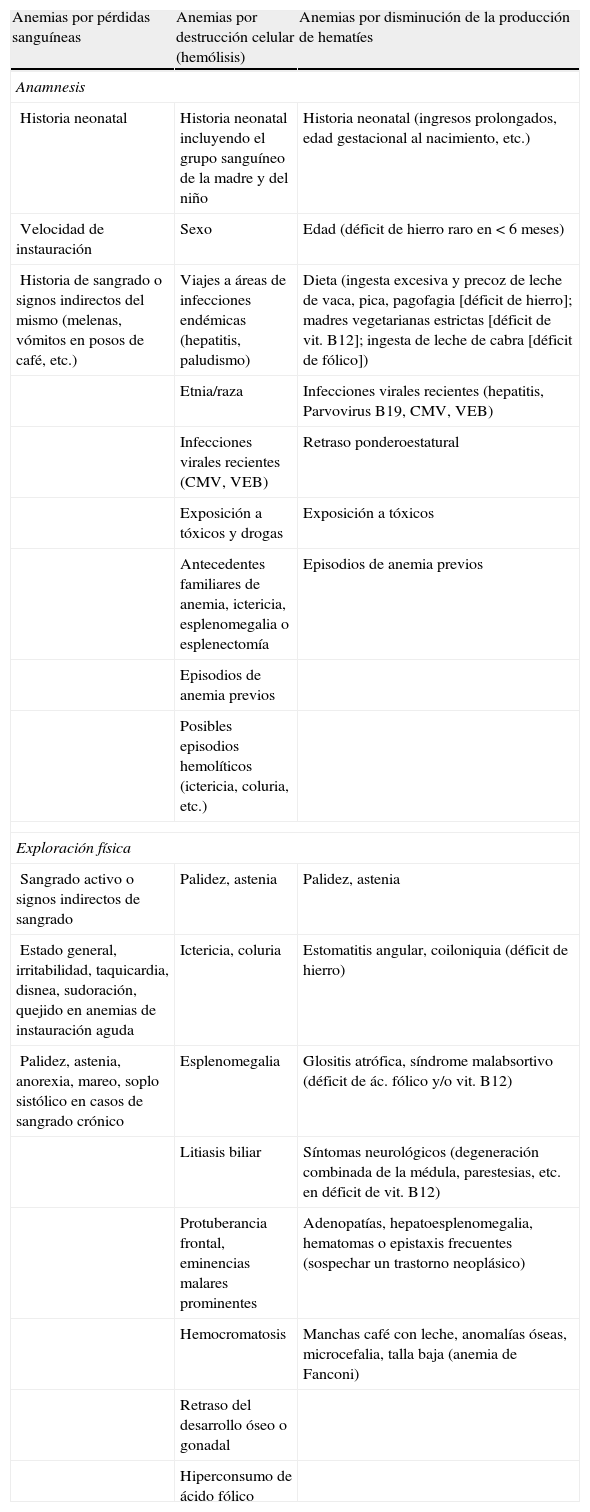
Una vez estabilizado el paciente, la aproximación diagnóstica debe basarse en la historia clínica detallada y la exploración física (tabla 2).
Anamnesis y exploración física
| Anemias por pérdidas sanguíneas | Anemias por destrucción celular (hemólisis) | Anemias por disminución de la producción de hematíes |
| Anamnesis | ||
| Historia neonatal | Historia neonatal incluyendo el grupo sanguíneo de la madre y del niño | Historia neonatal (ingresos prolongados, edad gestacional al nacimiento, etc.) |
| Velocidad de instauración | Sexo | Edad (déficit de hierro raro en < 6 meses) |
| Historia de sangrado o signos indirectos del mismo (melenas, vómitos en posos de café, etc.) | Viajes a áreas de infecciones endémicas (hepatitis, paludismo) | Dieta (ingesta excesiva y precoz de leche de vaca, pica, pagofagia [déficit de hierro]; madres vegetarianas estrictas [déficit de vit. B12]; ingesta de leche de cabra [déficit de fólico]) |
| Etnia/raza | Infecciones virales recientes (hepatitis, Parvovirus B19, CMV, VEB) | |
| Infecciones virales recientes (CMV, VEB) | Retraso ponderoestatural | |
| Exposición a tóxicos y drogas | Exposición a tóxicos | |
| Antecedentes familiares de anemia, ictericia, esplenomegalia o esplenectomía | Episodios de anemia previos | |
| Episodios de anemia previos | ||
| Posibles episodios hemolíticos (ictericia, coluria, etc.) | ||
| Exploración física | ||
| Sangrado activo o signos indirectos de sangrado | Palidez, astenia | Palidez, astenia |
| Estado general, irritabilidad, taquicardia, disnea, sudoración, quejido en anemias de instauración aguda | Ictericia, coluria | Estomatitis angular, coiloniquia (déficit de hierro) |
| Palidez, astenia, anorexia, mareo, soplo sistólico en casos de sangrado crónico | Esplenomegalia | Glositis atrófica, síndrome malabsortivo (déficit de ác. fólico y/o vit. B12) |
| Litiasis biliar | Síntomas neurológicos (degeneración combinada de la médula, parestesias, etc. en déficit de vit. B12) | |
| Protuberancia frontal, eminencias malares prominentes | Adenopatías, hepatoesplenomegalia, hematomas o epistaxis frecuentes (sospechar un trastorno neoplásico) | |
| Hemocromatosis | Manchas café con leche, anomalías óseas, microcefalia, talla baja (anemia de Fanconi) | |
| Retraso del desarrollo óseo o gonadal | ||
| Hiperconsumo de ácido fólico | ||
CMV: citomegalovirus; VEB: virus de Epstein-Barr.
Las manifestaciones clínicas de la anemia suelen ser inespecíficas. La palidez es el signo más común, evidente con valores de hemoglobina<9g/dl. Otras manifestaciones son astenia, letargia, anorexia, irritabilidad, soplo sistólico, datos de hemólisis, etc.5,6.
Las pruebas complementarias se realizarán en función de los datos obtenidos en la anamnesis y la exploración7,8.
En un primer estudio básico, realizaremos una analítica de sangre con hemograma que incluya índices eritrocitarios (VCM, HCM, CHCM, RDW), porcentaje de reticulocitos y frotis de sangre periférica. El valor de reticulocitos nos permitirá clasificar la anemia en arregenerativa o hiporregenerativa (reticulocitos<2%) e hiperregenerativa (> 2%) (algoritmo 1, fig. 1)1,9,10.
En función de la sospecha clínica, solicitaremos otras determinaciones: si sospechamos una anemia hemolítica, solicitaremos urea, creatinina, transaminasa glutamicoxalacética, bilirrubina, haptoglobina, LDH y test de Coombs; en el caso de sospechar un proceso neoplásico, LDH, ácido úrico, fósforo, sodio, potasio y cloro; gota gruesa en caso de sospechar paludismo, y reactantes de fase aguda si pensamos en un proceso inflamatorio agudo.
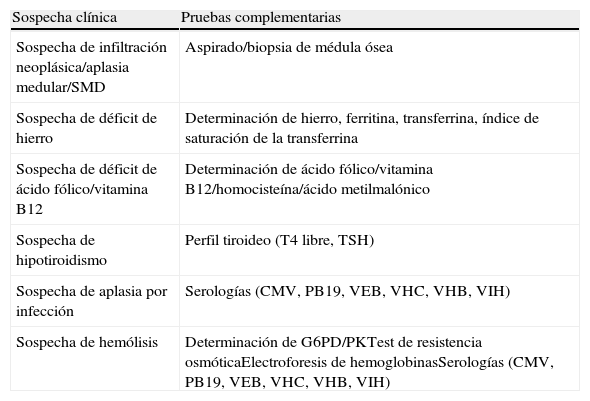
Se debe completar el estudio diagnóstico durante el ingreso o de forma ambulatoria siguiendo las indicaciones de la tabla 311,12.
Estudio diagnóstico posterior
| Sospecha clínica | Pruebas complementarias |
| Sospecha de infiltración neoplásica/aplasia medular/SMD | Aspirado/biopsia de médula ósea |
| Sospecha de déficit de hierro | Determinación de hierro, ferritina, transferrina, índice de saturación de la transferrina |
| Sospecha de déficit de ácido fólico/vitamina B12 | Determinación de ácido fólico/vitamina B12/homocisteína/ácido metilmalónico |
| Sospecha de hipotiroidismo | Perfil tiroideo (T4 libre, TSH) |
| Sospecha de aplasia por infección | Serologías (CMV, PB19, VEB, VHC, VHB, VIH) |
| Sospecha de hemólisis | Determinación de G6PD/PKTest de resistencia osmóticaElectroforesis de hemoglobinasSerologías (CMV, PB19, VEB, VHC, VHB, VIH) |
CMV: citomegalovirus; G6PD: glucosa 6-fosfato deshidrogenasa; PB19: Parvovirus B19; PK: piruvato cinasa; THS: hormona tiroestimulante; VEB: virus de Epstein-Barr; VHB: virus de la hepatitis B; VIH: virus de la inmunodeficiencia humana; VHC: virus de la hepatitis C.
En urgencias, la paciente presentaba regular estado general, palidez, fiebre y taquicardia. Pese a ello, no se estabilizó adecuadamente, puesto que no se siguió la secuencia ABCDE administrándose oxígeno de alto flujo, ni considerándose la necesidad de líquidos mientras se esperaba el resultado de la analítica sanguínea y las pruebas cruzadas.
En esta paciente se solicitaron todas las pruebas diagnósticas de entrada, sin realizar una aproximación diagnóstica inicial. La bioquímica se solicitó ante la sospecha de hemólisis o proceso neoplásico, al igual que el frotis de sangre periférica, el test de Coombs y el estudio específico de hemólisis. El estudio de hierro puede justificarse ante la anemia microcítica e hiporregenerativa. Sin embargo, la determinación de vitamina B12, ácido fólico y el perfil tiroideo no sería necesaria en un primer estudio diagnóstico desde el servicio de urgencias.
La presencia de datos clínicos y analíticos de hemólisis junto con test de Coombs directo negativo y esferocitos en sangre periférica debe orientarnos hacia una anemia hemolítica tipo esferocitosis hereditaria. Además, nuestra paciente presentaba un porcentaje de reticulocitos bajo, datos de infección e IgM positiva para Parvovirus B19 y virus de Epstein-Barr. Por todo ello, el diagnóstico final fue de crisis aplásica por ambos virus en una paciente con esferocitosis hereditaria13.
La esferocitosis hereditaria es la causa más frecuente de hemólisis hereditaria en individuos de raza blanca. Se trata de una alteración en genes que codifican para proteínas de membrana (anquirina, espectrina, banda 3, proteína 4.2), lo que conlleva a la ausencia de interacciones adecuadas del citoesqueleto con la bicapa lipídica, disminuyendo así la estabilidad de la membrana.
El hematíe adquiere una forma esferocítica que no es capaz de adaptarse a la microcirculación, quedando atrapado en los sinusoides esplénicos y destruyéndose en dicho órgano. Como consecuencia, un elevado porcentaje de pacientes presenta esplenomegalia. La esplenectomía mejora la anemia, disminuye el número de reticulocitos y el valor de la bilirrubina14.
El diagnóstico puede realizarse gracias a la historia familiar y la clínica junto con esplenomegalia y la analítica compatible. La presencia de esferocitos puede confirmarse con el test de fragilidad osmótica. La citometría de flujo con eosin-5-maleimide o el test de autohemólisis son otros métodos diagnósticos15,16.
Las crisis aplásicas suelen ser secundarias a infecciones víricas, principalmente a Parvovirus B19. La insuficiencia eritroide medular puede causar rápidamente una anemia intensa y clínica de insuficiencia cardíaca, hipoxia y fallo cardiovascular. También pueden disminuir las demás series. Clínicamente, se manifiesta con palidez, astenia, anorexia, irritabilidad y taquicardia. Pueden referir el antecedente de un episodio febril con dolor abdominal, diarrea, artralgias, mal estado general y exantema (aunque a veces la infección es subclínica). El episodio revierte espontáneamente en 2-14 días17,18.
El virus de Epstein-Barr puede compartir el mismo mecanismo con el Parvovirus B19 e inducir una aplasia de médula ósea, aunque la patogénesis de este fenómeno es aún desconocida. Existen pocos casos descritos de coinfección por virus de Epstein-Barr y Parvovirus B19 en pacientes con esferocitosis hereditaria, por lo que la clínica de dicha coinfección no es muy conocida, habiéndose descrito desde infecciones asintomáticas hasta la completa supresión de médula ósea19.
El tratamiento de estos episodios es sintomático, recurriendo a la transfusión de hematíes en caso de indicación.
ConclusionesAnte un paciente letárgico, pálido, con irritabilidad, pobre ingesta oral, taquicardia o datos de hemólisis debemos sospechar un síndrome anémico.
En urgencias, debemos actuar siguiendo la secuencia ABCDE1. Se realizará una primera aproximación diagnóstica con la anamnesis, la exploración y el hemograma con reticulocitos, frotis sanguíneo y estudio bioquímico de hemólisis si se sospecha clínicamente. Las pruebas etiológicas específicas deben basarse en este primer estudio.
En los pacientes que precisen transfusión de sangre en la estabilización inicial, se tendrá en cuenta que esto puede retrasar la realización de pruebas complementarias etiológicas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
