
Mineralización ósea y marcadores genéticos en niños afectos de trastornos de la conducta alimentaria*
A. Díez, C. Azcona, A. Patiño, C. Soutullo, J. Gimeno, E. Sotillo, L. Alonso, M. Portas e I. Landecho
Unidad de Endocrinología Pediátrica. Departamento de Pediatría. Clínica Universitaria. Pamplona.
La osteopenia en los trastornos de la conducta alimentaria (TCA), principalmente en la anorexia nerviosa, es una complicación de gran importancia debido al posterior riesgo de desarrollar osteoporosis y fracturas.
Objetivos.1) valorar la mineralización ósea (MO) mediante dos técnicas de medición diferentes: DEXA y osteosonografía digital; 2) analizar algunos de los marcadores genéticos que han sido previamente relacionados con la masa ósea, y 3) cuantificar y valorar la ingesta diaria de calcio y vitamina D.
Se incluyen 61 pacientes afectos de trastornos de la conducta alimentaria (58 mujeres y 3 varones) con una edad media de 18,4 años (IC 95%: 17,3; 19,5). Se realiza tipaje genético en 142 individuos sanos como grupo control. Las determinaciones de MO se hacen mediante DEXA (Dual-Energy-X ray Absortiometry, HOLOGIC QDR 4500 Elite, Hologic, Woltham, MA) y osteosonografía (equipo DBM Sonic-1200), en la metáfisis distal de las falanges proximales del 2º al 5º dedos de la mano no dominante.Se extrae ADN de sangre periférica y se obtienen los genotipos para los marcadores FokI, ApaI, TaqI del gen del receptor de la vitamina D (VDR); XbaI y PvuII del gen del receptor de estrógeno (ER) y MscI del gen del colágeno I*I (ColI*I). Los alelos para cada uno de los marcadores se determinan mediante RFLP (restriction fragment length polymorphisms), asignándose la letra mayúscula a la forma no cortada (sin diana) y la minúscula a la cortada (con diana).
El tiempo medio de evolución de la enfermedad es de 3,7 años (IC 95%: 2,8; -4,7). El índice de masa corporal es de -1,4 SDS (IC 95%: -1,9; 1,0). La MO determinada por DEXA (n = 24) muestra un valor medio de -0,9 SDS (IC 95%: -1.2; -0,5) y la obtenida mediante osteosonografía (n = 42) es de -1,8 SDS (IC 95%: -2,5; -1,2). El tiempo de amenorrea se correlaciona con el peso (r = -0,36;p < 0,05), y con los valores de MO determinados tanto por DEXA como por osteosonografía (r = -0,50; p < 0,05). El peso se correlaciona de forma positiva con la MO determinada por DEXA (r = 0,46; p < 0,05) y sin embargo la correlación es inversa si la MO es determinada mediante osteosonografía (r = -0,73; p < 0,05). No se detectaron diferencias estadísticamente significativas en la distribución de los genotipos entre la población con TCA y los controles; sin embargo, parece existir una tendencia, en los pacientes estudiados afectos de TCA, a presentar con mayor frecuencia los genotipos heterocigotos Xx y Pp del gen ER. En pacientes afectos de TCA, la presencia del alelo F, tanto en su forma homocigota FF como en la heterocigota Ff, existe una tendencia a tener mayores valores de DMO lumbar. La ingesta media diaria de calcio de los pacientes es de 656,2 mg (IC 95%: 421; 890) y de vitamina D es 4,5 mcg (IC 95%: 3,1; 5,9); ambas son inferiores a las recomendaciones diarias para la edad (calcio: Z = -2,8; p < 0,05, y vitamina D: Z = -3,2; p < 0,001) (RDA 1989).
Los pacientes afectos de TCA presentan una MO disminuida determinada mediante dos métodos de medición: osteosonografía digital y DEXA. No se observan diferencias en la distribución de los genotipos estudiados entre la población con TCA y los controles. La ingesta diaria de vitamina D y calcio se encuentran por debajo de las recomendaciones diarias.
*Premio Pharmacia a la mejor comunicación oral.
El tratamiento con metformina normaliza la función ovulatoria en adolescentes con hiperandrogenismo ovárico e hiperinsulinismo
L. Ibáñez1, C. Valls1, A. Ferrer1, F. de Zegher2, N. Potau3, M.V. Marcos4 y F. Rodríguez-Hierro1
1Hospital Sant Joan de Déu, Universidad de Barcelona, 2Universidad de Lovaina, Bélgica. 3Hospital Materno-Infantil Vall d'Hebron, Universidad Autónoma de Barcelona. 4Consorci Hospitalari de Terrassa.
Las niñas con pubarquia precoz (PP) presentan un riesgo elevado de desarrollar disfunción ovulatoria, hiperinsulinismo e hiperandrogenismo ovárico en la adolescencia. El tratamiento con metformina, un agente sensibilizante de la insulina, disminuye el hiperinsulinismo y el hiperandrogenismo ovárico; sin embargo se desconocen los efectos sobre la función ovulatoria.
Hemos estudiado si la administración de metformina (1.275 mg/d durante 6 meses) es capaz de inducir la ovulación en 18 adolescentes no obesas (edad: 16,5 ± 0,4 años; índice de masa corporal: 21,4 ± 0,6 Kg/m2) con ciclos anovulatorios e historia de PP. Todas las pacientes presentaban hirsutismo (score de Ferriman y Gallwey 15,4 ± 0,8), hiperinsulinismo (pico de insulina > 150 µU/mL durante la sobrecarga oral de glucosa), dislipemia e hiperandrogenismo ovárico (diagnóstico mediante test de agonistas de GnRH); la menarquia se había producido al menos tres años antes (rango: 3-7 años). Antes de iniciar el tratamiento, se comprobó en dos ocasiones (meses -3 y -1), que los ciclos eran anovulatorios en todas las adolescentes, mediante la medición durante 4 semanas consecutivas de las cifras de progesterona sérica; el porcentaje de pacientes con ciclos ovulatorios y anovulatorios se evaluó de manera similar después de 2, 4 y 6 meses del tratamiento con metformina.
Antes de iniciar el tratamiento con metformina, los niveles de progesterona sérica fueron < 4 ng/mL en todas las determinaciones. A los 2 meses de iniciado el tratamiento, sólo una de las pacientes presentó ciclos ovulatorios (niveles de progesterona sérica > 8 ng/mL). Sin embargo, el número de pacientes con ciclos ovulatorios fue de 7/18 (39%) a los 4 meses, y de 15/18 (83%) a los 6 meses de administrar metformina.
El tratamiento con metformina se acompañó de una reducción del grado de hirsutismo, de las concentraciones séricas de insulina y andrógenos, de un aumento de las cifras de sex hormone-binding globulin (p < 0,01 para todos los parámetros), y de una normalización del perfil lipídico.
En conclusión, la administración de metformina es capaz de normalizar la función ovulatoria en adolescentes no obesas con historia de PP que desarrollan hiperinsulinismo e hiperandrogenismo ovárico. La indicación de los agentes sensibilizantes de la insulina en el tratamiento del hiperinsulinismo asociado a hiperandrogenismo ovárico en la adolescencia precisa, sin embargo, de estudios randomizados que demuestren la eficacia y tolerancia a largo plazo de estos fármacos.
Bajo peso al nacer, hiperinsulinismo y pubarquia precoz en niñas: relación con los polimorfismos de los VNTR del gen de la insulina
L. Ibáñez1, D. Dunger2, F. de Zegher3, A. Ferrer1, N. Potau4, M.V. Marcos5 y F. Rodríguez-Hierro1
1Hospital Sant Joan de Déu, Universidad de Barcelona. 2Universidad de Cambridge, Reino Unido. 3Universidad de Lovaina, Bélgica.4Hospital Materno-Infantil Vall d'Hebron, Universidad Autónoma de Barcelona.5Consorci Hospitalari de Terrassa.
En diversos grupos poblacionales se han descrito asociaciones entre bajo peso al nacer, hiperinsulinismo, y otros factores de riesgo para el desarrollo de enfermedad en la edad adulta. Hemos estudiado si en las niñas con pubarquia precoz (PP), estas asociaciones podrían estar en parte determinadas por determinados polimorfismos en los "variable number of tandem repeats" del gen de la insulina (INS VNTR), que juegan un papel fundamental en la regulación de la expresión del gen de la insulina.
En 124 niñas con historia de PP se determinó la sensibilidad a la insulina a partir de las cifras basales de glucosa e insulina, las concentraciones medias de insulina (MSI) en respuesta a una sobrecarga oral de glucosa, el perfil lipídico, las cifras de la proteína transportadora de las hormonas sexuales (SHBG), y en las pacientes postpuberales, la respuesta de la 17-hidroxiprogesterona (17-OHP) ovárica a la estimulación con un análogo de GnRH. En las pacientes y en un grupo control (n = 140) de edad e índice de masa corporal similar, se estudió el genotipo de los INS VNTR, específicamente las variables alélicas de clase I y clase III.
En las niñas con PP, el peso al nacer presentó una correlación inversa con el MSI (r = 0,34, p < 0,0005), y con las cifras de colesterol total (r = 0,66, p < 0,0005), y de LDL-colesterol (r = 0,43, p < 0,0005). La distribución de los genotipos de los INS VNTR fue similar en las pacientes y controles. En las pacientes con uno o dos alelos de clase I (I/*), el peso al nacer fue significativamente inferior al de las pacientes con genotipo III/III (p = 0,004). Las niñas con PP y genotipos I/I y I/III, presentaron cifras más elevadas de MSI y menor sensibilidad a la insulina que las niñas con genotipo III/III. El análisis de regresión logística mostró efectos adversos aditivos del bajo peso al nacer y del genotipo I/* sobre el MSI y las cifras de LDL-colesterol. El genotipo de los INS VNTR fue el máximo determinante de la sensibilidad a la insulina y de las cifras de SHBG, mientras que las cifras de 17-OHP post-análogo y los niveles de triglicéridos sólo se correlacionaron con el peso al nacer.
En las niñas con PP, el riesgo de desarrollar hiperinsulinismo y endocrinopatías en la adolescencia parece estar co-determinado por la existencia de un bajo peso al nacer y de al menos un alelo de clase I en los INS VNTR. Similares interacciones entre genotipo e hipocrecimiento prenatal podrían explicar también otras asociaciones entre bajo peso al nacer y enfermedades en la edad adulta.
Densidad mineral ósea (DMO) a nivel lumbar en recién nacidos prematuros. Estudio transversal y longitudinal
D. Yeste*, M. Clemente*, J. Almar**, M. Gussinyé*, G. Peguero** y A. Carrascosa*
*Servicio de Endocrinología Pediátrica. **Servicio de Neonatología. Hospital Materno-Infantil Vall d'Hebrón. Barcelona.
Fundamento. El máximo depósito de sales minerales del esqueleto fetal se adquiere en el 3° trimestre del embarazo, produciéndose el pico máximo de acreción ósea intrauterino entre las 33 y 36 semanas de gestación. El RNPT nace con una mineralización deficiente, constituyendo una población de riesgo para desarrollar osteopenia y raquitismo. Con objeto de conocer la incidencia de osteopenia del RNPT en la vida postnatal y su evolución posterior se ha estudiado de forma transversal (n = 80) y longitudinal (n = 31) la densidad mineral ósea (DMO) en una población de RNPT a las 46 semanas posconcepción, a la edad de 1 año y de 2 años mediante técnicas no invasivas de densitometría radiológica de doble energía (DEXA).
Pacientes y métodos. La DMO del esqueleto fue valorada a nivel de la columna lumbar, L2-L4, mediante DEXA (modelo Lunar DPX) en 80 RNPT (41 niños y 39 niñas) de edad gestacional comprendida entre las 24 y 35 semanas de gestación (media: 31,2 ± 2,5), y los valores obtenidos comparados con los de una población de recién nacidos a término normales de idéntica edad cronológica (n = 84). El aporte medio estimado de calcio, fósforo y vitamina D durante el tiempo de hospitalización fue de 130 mg/kg/día, 72 mg/kg/día y 450 UI/día respectivamente.
Resultados. Los valores de DMO se recuperan progresivamente desde el nacimiento hasta la edad de 2 años. El valor de DMO a la edad posconcepcional de 46 semanas es de 0,139 ± 0,06 g/cm2 (-2,4 ± 1,4 DS) y muestra una correlación positiva y estadísticamente significativa con la edad gestacional (r = 0,63, p < 0,001, n = 55), el peso del nacimiento (r = 0,31, p = 0,02, n = 55) y la talla al nacer (r = 0,35, p = 0,01, n = 47). La DMO a la edad de 1 año es de: 0,270 g/cm2 (-1,0 ± 1,0 DS) y a la edad de 2 años: 0,410 g/cm2 (-0,08 ± 1,0 DS). El porcentaje de RNPT con valores de DMO inferiores a -2 DS se reduce desde el 56% a la edad de 46 semanas de vida posconcepcional, al 19% a la edad de 1 año, y es del 2% a la edad de 2 años. No se observan diferencias estadísticamente significativas en el valor de DMO alcanzado a la edad de 2 años en función de la edad gestacional en ambos estudios.
Conclusiones. Nuestros datos muestran que la DMO a nivel lumbar de los RNPT, evaluada mediante DEXA, se recupera de forma progresiva, alcanzando valores similares de DMO a los de los recién nacidos a término a la edad de 2 años e independientemente de la edad gestacional.
Retinopatía en enfermos afectos de diabetes tipo I: disminución de la incidencia con el uso de dos dosis diarias de insulina
C.W. Ruiz, A. Edo, M. Gussinyer, M.A. Albisu, D. Yeste, E. Vicens, E. Armengol, P. Olivan y A. Carrascosa
Servicio de Endocrinología Pediátrica del Hospital Materno-Infantil Vall d'Hebrón. Barcelona.
Objetivo. Describir la incidencia de retinopatía entre la población de enfermos con diabetes mellitus tipo 1 (DM-I) controlados en nuestro centro. Asimismo. se intentará relacionar la variación experimentada en la misma, respecto a estudios previos, con la introducción de una segunda dosis de insulina diaria a partir de 1980 (y 3 o más dosis a partir de 1990).
Material y métodos. Estudio descriptivo retrospectivo donde se han incluido los enfermos con DM-I que debutaron entre 1980 y 1990, y que han sido seguidos un mínimo de 10 años en nuestro hospital. La exploración oftalmológica se realizó mediante oftalmoscopía tridimensional, con controles anuales a partir del quinto año de evolución de la diabetes. Los resultados de las variables cuantitativas se expresan en forma de media y rango. El análisis estadístico se ha realizado aplicando la prueba de Mann-Whitney.
Resultados. Se han estudiado 176 pacientes, aunque sólo en 80 de ellos (45,45%) el seguimiento ha sido igual o superior a 10 años. De éstos, 11 (13,75%) han presentado signos compatibles con retinopatía diabética tras una media de 11,11 añs de evolución de la diabetes (7,42-18,5), y siempre una vez finalizado el desarrollo puberal. Todos los casos se informaron como retinopatía incipiente, en forma exclusivamente de microaneurismas sin progresión en sucesivos controles. En 10 de los 11 casos (90,91%) la retinopatía se presentó asociada a hipertensión, nefropatía o neuropatía periférica subclínica. En nuestro hospital, a partir de 1980 se pasó de una a dos administraciones de insulina al día; a pesar de ello, 4 de los 11 enfermos con retinopatía (36,36%) estuvieron entre 1 y 2 años recibiendo una sola dosis desde el debut de la diabetes. En el momento del diagnóstico de la retinopatía todos los pacientes recibían 2 o 3 dosis de insulina diarias. El valor medio de HbAlc entre los afectos de retinopatía fue de 8,25% (7,10-9,20), mientras que en el grupo de diabéticos sin retinopatía este valor fue de 7,56% (5,26-11,20); no obstante, esta diferencia no posee suficiente significación estadística (p = 0,061). La diferencia entre ambos grupos tampoco es estadísticamente significativa en cuanto a la edad de debut de la diabetes (p = 0,91).
Conclusiones. Comparando con el estudio previo, presentado el año 1992, con 88 pacientes diabéticos que debutaron entre 1966 y 1980 y que fueron seguidos al menos l0 años y donde un 50% presentaba retinopatía, la disminución importante de la incidencia de esta complicación a un 13,75% se puede atribuir al mejor control metabólico conseguido gracias a la administración de dos dosis de insulina en lugar de una como previamente. Además, la ausencia de diferencia entre los valores de HbAlc entre los enfermos con y sin retinopatía establece que, dentro de un control metabólico aceptablemente bueno, la aparición de retinopatía tiene una base predominantemente idiosincrática. Se deberá esperar para saber si el cambio a 3 o más dosis, generalizado a partir de 1990, se traduce en unas cifras de retinopatía, si cabe, inferiores.
Agradecimiento: Servei de Documentació Clínica i Arxiu del Hospital Materno-Infantil Vall d'Hebrón.
Área grasa y muscular braquiales en población infantil
I. Rica*, E. Sánchez** y B. Sobradillo**
Endocrinología Pediátrica. Hospitales de Cruces* y de Basurto**. Bizkaia.
La medición antropométrica del antebrazo es de gran utilidad para la valoración nutricional. Gurney y Jelliffe en 1973 establecieron una ecuación para estimar el "área grasa y muscular braquiales", en función de las medidas del perímetro braquial y pliegue tricipital. El empleo de estas variables sería útil en el control de trastornos nutricionales como la obesidad exógena y la anorexia nervosa.
Objetivos.1) Estandarización en la población de Bilbao de 0 a 14 años, de las variables área grasa (AGB) y muscular (AMB) braquiales, estratificados por edad y sexo. 2) Comparación entre sexos de dichas variables así como con los resultados publicados en otras poblaciones.
Sujetos y metodología. El diseño del estudio fue longitudinal-mixto, reclutándose 3 cohortes de niños/as (recién nacidos, 5 años de edad y 9 años) seleccionados al azar de la población de Bilbao, en el periodo de 1978 a 1980 que incluyeron un total de 1846. Las mediciones antropométricas se realizaron semestralmente durante 6 años. Las curvas referentes al pliegue tricipital (PT) y perímetro braquial (PB) han sido publicadas previamente dentro del estudio de crecimiento de Bilbao. Hemos calculado el AGB y AMB empleando las ecuaciones descritas por Gurney y Jelliffe. Posteriormente se han calculado los estadísticos: media y desviación estándar para cada edad y sexo. Las curvas se han construido tras realizar una transformación logarítmica de los datos representando los percentiles 3, 25, 50, 75 y 97. Hemos comparado nuestros resultados con otros estándares previamente publicados mediante comparación de medias.
Resultados.1) Curvas de estandarización.
2) El AGB es superior en las niñas a partir de los 4 años de edad; el AMB es superior en los niños a partir de los 10 años. Los valores del AGB y AMB difieren con los estándares publicados para población americana y suiza, siendo similares a los datos de población preescolar de Madrid.
Conclusiones.1)Se ha estandarizado el AGB y AMB en población infantil de 0 a 14 años; el comportamiento de estos indicadores varía en función del sexo. 2) Existen diferencias con los datos de otras poblaciones por lo que es necesario disponer de valores de referencia propios para su aplicación en el control de trastornos de la nutrición.
Relación entre los polimorfismos del gen del receptor de la vitamina D (VDR) en exón 2 e intrón 8 y la mineralización ósea en la diabetes mellitus tipo 1 (DM1)
G. Martí, C. Esteban, M. Fernández, M. Gussinyé, D. Yeste, M. Albisu, E. Vicens, A. Carrascosa y L. Audí
Hospital Materno-Infantil Vall d'Hebrón. Barcelona.
Introducción. El gen VDR se ha descrito como uno de los factores genéticos implicados en la regulación de la mineralización ósea. La existencia en la población pediátrica de una serie del enfermedades que predisponen a la osteopenia nos motiva a estudiar los polimorfismos del gen VDR como posibles predictores del grado de mineralización ósea.
Objetivo. Relacionar los dos polimorfismos del gen VDR situados en el exón 2 (Fokl) y en el intrón 8 (BsmI) en pacientes afectos de DM1 con el estado de mineralización ósea a lo largo de su enfermedad.
Material y métodos. Determinación en 74 pacientes DM1 (X ± DE 17,1 ± 4,4 años) de 8,1 ± 5 años de edad al inicio y 8,9 ± 6,1 años de evolución (tabla I) de los polimorfismos BsmI (BB, Bb, bb) y Fokl (FF, Ff, ff), previa obtención de DNA genómico a partir de sangre periférica, amplificación mediante la reacción en cadena de la polimerasa (PCR) de las secuencias de DNA que contienen la diana para BsmI y Fokl, digestión con la enzima de restricción específica y/o secuenciación del segmento de DNA. Cuantificación del contenido mineral óseo a nivel lumbar (L2-L4) mediante densitometría ósea (DMO). Estudio del apareamiento de los genotipos de cada polimorfismo y la combinación de ambos con el valor de mineralización ósea y el tiempo de evolución de la enfermedad con F-test, así como la distribución de genotipos y sus combinaciones en relación al estado de mineralización mediante el test X2.
Resultados.

La clasificación de las DMO según el grado de mineralización ósea (tabla 1) permite observar una mayor presencia del genotipo bb en el grupo de pacientes osteoporóticos y del alelo fen los grupos de osteopénicos y osteoporóticos.
Hasta los 5 años de evolución (n = 22) la DMO (-0,696 ± 1,032) muestra diferencias significativas (p = 0,047) para los genotipos BsmI (bb = -0,956, Rb = -0,633, RR = -0,017), así como para los genotipos Fokl (p = 0,047) (ff = -0,870, Ff = -0,787, FF = -0,626).
Tras seleccionar el grupo de pacientes (n = 45) que finalizaron la pubertad (Tanner G4 o S4) se observa que la presencia de 2 alelos ff para el polimorfismo Fokl empeora la DMO, así el genotipo Bbff presenta una peor mineralización comparada con la de los genotipos bbFf (p = 0,01), BbFF (p = 0,02) y bbFF (P = 0,02).
Conclusiones. Los polimorfismos del gen VDR parecen influir sobre la masa ósea en pacientes con DM1: El genotipo bb y el alelo f condicionan una menor mineralización ósea.
Evidencia de incremento en la apoptosis celular en la hipófisis anterior de ratas con diabetes mellitus experimental mal controlada
A.I. Arroba, L.M. Frago, J. Arente y J.A. Chowen
Hospital Universitario Niño Jesús. Endocrinología y Laboratorio de Investigación. Madrid.
Introducción. El mal control metabólico de la diabetes mellitus conduce a complicaciones multisistémicas, pudiendo afectarse, además de la retina, la función renal y el sistema nervioso central y periférico, la hipófisis. La afectación de ésta puede alterar la secreción de las hormonas en esta glándula. No obstante, los mecanismos íntimos responsables de dichas alteraciones no están esclarecidos. Como quiera que la hipófisis anterior está sometida a cambios en proliferación celular y apoptosis incluso en la época adulta, su composición celular es susceptible de modificarse en función del ambiente local.
Objetivo. Investigar el efecto de la diabetes crónica sobre la apoptosis y composición celular en la glándula hipófisis.
Diseño experimental y metodología. Se emplearon ratas Wistar macho adultas a las que se inyectó estreptozotocina para inducir la diabetes. El grupo control recibió vehículo. Un grupo de ratas diabéticas se trataron con dos dosis/día de insulina NPH para mantener niveles de glucemia próximos ala. normalidad {bien controladas); mientras un segundo grupo recibió una única dosis de insulina NPH para mantener viables los animales (mal controladas). Estos animales se sacrificaron después de 3 semanas de padecer diabetes. Un tercer grupo de ratas diabéticas no recibieron insulina, sacrificándose después de dos meses (diabetes a largo plazo). Tras el sacrificio de los animales, se extirparon sus hipófisis; éstas se congelaron inmediatamente y, posteriormente, se realizaron cortes en criostato (12 µm). Se efectuaron reacciones TUNEL para detectar la presencia de cuerpos apoptóticos, e hibridación in situ no radioactiva para prolactina y hormona de crecimiento para determinar la densidad de lactotropas y somatotropas.
Resultados. Se detectaron escasos cuerpos apoptóticos en el tejido hipofisario de las ratas control y de las ratas diabéticas bien controladas. Por el contrario, los animales diabéticos mal controlados presentaban significativamente más. Finalmente, las ratas con diabetes a largo plazo, mostraban la mayor densidad de formaciones apoptóticas. Aunque no se apreció ninguna diferencia significativa en la densidad de somatotropas, las ratas con diabetes a largo plazo exhibían una disminución significativa en la densidad de lactotropas.
Conclusión. La diabetes mellitus mal controlada puede conllevar un incremento en apoptosis en la hipófisis anterior, siendo las células lactotropas más susceptibles a este fenómeno.
Pubarquia precoz e hiperandrogenismo funcional ovárico en adolescentes. Descripción de una serie
M. Gómez-Balaguer, E. Solá, S. Garzón, S. García, H. Peña, M.L. Muñoz, C. Morillas, D. Lorente y A. Hernández
Servicio Endocrinología. Hospital Universitario Doctor Peset. Valencia.
La pubarquia precoz (PP) o exagerada, definida como la aparición de vello pubiano por debajo de los 8 años, es un buen marcador clínico de hiperandrogenismo funcional ovárico (FOH). La prevalencia del FOH, asociando dislipemia e hiperinsulinismo, entre las adolescentes con historia de PP está aumentada, pero se desconoce su número. Se presentan los datos descriptivos de una serie de FOH, con especial referencia a la presencia de PP.
Han sido estudiadas 202 mujeres por clínica de hirsutismo y/o alteraciones del ciclo menstrual. Entre los datos de la anamnesis, se recogieron los referentes a la edad de la pubarquia, la talla y el peso, calculándose el índice de masa corporal (IMC). Todas las pacientes fueron sometidas a un protocolo uniforme de diagnóstico, mediante la determinación de la respuesta de 17-hidroxiprogesterona (17-OH-Pg) al estímulo de GnRH bajo frenación con dexametasona. Cuando las basales de 17-OH-Pg eran elevadas, se practicó un test de ACTH para 17-OH-Pg para descartar déficit de 21-hidroxilasa. Se determinaron además S-DHEA, androstendiona, cociente LH/FSH, SHBG, testosterona total, índice androgénico libre (FAI), perfil lipídico, insulina y péptido C. Las pacientes con hiperrespuesta de 17-OH-Pg tras estímulo con GnRH fueron diagnosticadas de FOH. Estas pacientes se compararon con un grupo control de 102 pacientes de igual rango de edad diagnosticadas de hirsutismo idiopático por medio del mismo protocolo diagnóstico.
Resultados. Se diagnosticaron 146 FOH (rango de edad 13-24 años), de las cuales 14 referían historia de PP (pubarquia entre los 6,2 y los 7,4 años), lo que supuso un 9,5% del total de FOH. En el grupo control, sólo 2 pacientes referían PP, lo que supone un 1,9%. Del conjunto de pacientes con FOH, el 4% presentaban un IMC mayor de 40 kg/m2 (obesidad mórbida), el 12% un IMC de 30 a 40 kg/m2 (obesidad moderada-severa), el 16% un IMC de 25 a 30 kg/m2 (sobrepeso), y el 24% un IMC entre 15 y 20 kg/m2 (delgadez). Las 14 pacientes con FOH e historia de PP presentaban IMC entre 15 y 20 kg/m2. No hubo diferencias estadísticamente significativas entre las pruebas hormonales del grupo de pacientes con FOH e historia previa de PP, y las del grupo de pacientes con FOH sin PP (respuesta a GnRH de 17-OH-Pg, S-DHEA, androstendiona, cociente LH/FSH, SHBG, testosterona total, FAI, insulina, péptido C, perfil lipídico).
Conclusión. Observamos una mayor presencia de PP en las pacientes con FOH que en las pacientes con hirsutismo idiopático. Curiosamente, el único parámetro que diferenció a las pacientes con FOH y PP de las pacientes con FOH sin PP fue su menor IMC.
Pubarquia precoz. Un análisis descriptivo de 154 casos
M.A. Fuentes, A. Copil, M. Gussinyé, D. Yeste, A. Albisu, E. Vicens y A. Carrascosa
Hospital Materno-Infantil Vall d'Hebrón. Barcelona.
Introducción. La pubarquia precoz es una patología prevalente que diversos estudios han asociado con pubertad adelantada, con mayor o menor repercusión en la talla final, con hiperinsulinismo, hiperandrogenismo ovárico, y bajo peso al nacimiento. En este estudio descriptivo hemos analizado de forma retrospectiva las pacientes controladas en nuestro hospital con objeto de dilucidar algunas de estas asociaciones avaladas por la literatura médica.
Material y métodos. Se han revisado de forma retrospectiva 154 casos (125 de sexo femenino y 29 de sexo masculino) que había iniciado la pubarquia antes de los 9 años de edad en niños y antes de los 8 años de edad en niñas, sin otros signos de progresión puberal asociados. Del total de 154 casos, en el momento del análisis 106 (68,8%) habían iniciado el desarrollo puberal y 51 (33,7%) habían alcanzado la talla adulta.
Resultados. Se pudo disponer del peso al nacimiento en 123 casos (80% de la muestra). Diez fueron prematuros ( < 37 SG) y presentaron todos un peso adecuado a su edad gestacional. De los restantes 113 recién nacidos a término, únicamente 7 casos (6%) tenían al nacer un peso menor de 2.500 g. La distribución percentilada de los pesos al nacimiento de nuestra muestra es: p - 3%: 2.005 g; p - 10%: 2.500 g; p - 25%: 2.850 g; p - 50%: 3.200 g; p - 75%: 3.500 g; p - 90%: 3.750 g y p - 97%: 4.116 g. Esta distribución percentilada es similar a la de los patrones normales de peso al nacimiento de la población normal de referencia.
La incidencia de pubertad precoz (aparición de desarrollo puberal antes de los 8 años en niñas y de los 9 años en niños), o adelantada (entre los 8 y los l0 años en niñas; entre los 9 y los 11 en niños), en nuestra muestra fue: a) sexo masculino cinco casos (26%) de pubertad adelantada y ninguno de pubertad precoz; b) sexo femenino 46 casos (53%) de pubertad adelantada y 2 casos (2,3%) de pubertad precoz. De los 51 casos en que se pudo disponer de talla final, sólo hubo 8 (15,5%) en que ésta fue inferior a la talla diana calculada en función de las tallas de los progenitores.
Discusión yconclusiones. A la vista de los resultados expuestos, podemos concluir que en nuestra muestra la pubarquia precoz no se asocia con una mayor incidencia de bajo peso al nacimiento que la de la población normal; que sí parece asociar un mayor riesgo de pubertad adelantada (sobre todo en el sexo femenino); y que este último hecho, sin embargo, no conlleva un efecto negativo sobre la talla final alcanzada.
Agradecimiento al "Servei de documentació clínica i arxiu"
Defectos moleculares en el gen CYP21 en niñas con pubarquia prematura
I. Eduardo1, L. Ibañez2, S. Riqué1, M. Gussinyé1, E. Vicens1, M.V. Marcos3, A. Carrascosa1 y N. Potau1
1Laboratorio Hormonal Hospital Materno-Infantil Vall d'Hebron. 2Unidad de Endocrinología Pediátrica Sant Joan de Deu. 3Hospital de Terrassa.
La pubarquia prematura (PP) se define como la aparición de vello pubiano ante de los 8 años en la niña y de los 9 años en el niño. La PP se debe a una maduración precoz y aislada de la glándula suprarrenal (adrenárquia precoz). La prevalencia de formas no clásicas de hiperplasia suprarrenal congénita (HSC) en niñas con PP es del 7-10%.
El objetivo del estudio es analizar la presencia de alelos mutados en el gen CYP21 en 60 niñas con PP y 35 niñas como controles sanos. En las niñas con PP se había descartado la presencia de una forma no clásica de HSC mediante un test del ACTH.
Después de una selección inicial del gen frente al pseudogen por PCR, se estudiaron por el método alelo-específico 14 mutaciones conocidas en el gen CYP21. Se identificaron como heterozigotos simples 14 niñas con PP y 8 controles, lo que representa una frecuencia del 23% para ambos grupos.
Las mutaciones encontradas se especifican en la tabla:

Los valores de 17-hidroxiprogesterona post-ACTH fueron similares en las portadoras y en las no portadoras (437,6 ± 398,0 ng/dl vs 268,0 ± 141,9 ng/dl). Tampoco hubo diferencias en la respuesta insulínica después de una sobrecarga oral de glucosa.
En conclusión en las niñas con PP no esta aumentada la presencia de portadoras de alelos mutados del gen CYP21 en relación con la población control estudiada. No parece existir relación entre la condición de portadoras y su estado endocrino-metabólico.
Ritmos circadianos de cortisol, testosterona y 17-OHP en distintas pautas terapéuticas en hiperplasia adrenal congénita por deficiencia
de 21-hidroxilasa
A. Rodríguez*, L. García*, A. Tejedor**, O. Serrano*, J.R. Arnao* y M.D.R. Arnao*
*Unidad de Metabolismo y Desarrollo. **Servicio de Nefrología. Hospital General Universiatrio Gregorio Marañón. Madrid.
Objetivos. Analizar la influencia del fraccionamiento en distintos horarios de una misma dosis total diaria de hidrocortisona oral (HC), en pacientes pediátricos con. deficiencia de 21-hidroxilasa (21-OHD).
Pacientes y métodos. 10 pacientes con 21-OHD, 8 niñas con edades comprendidas entre 3 años y 2 meses y 16 años; 2 varones de 6 años y 2 meses y 6 años y 3 meses. Todos en tratamiento con hidrocortisona oral (dosis: 8,69-27,55 mg/m2/día) y 9-alfa-fluorhidrocortisona (dosis: 10,75-43,85 mcg/m2/día).
Protocolo de estudio: Dosis diaria total constante de HC, fraccionada según 3 pautas en cada paciente, mantenidas durante 30 días. Pauta A: 1-0-0. Pauta B: 0-0-1. Pauta C: 2/3-0-1/3, donde los guarismos representan la fracción de la dosis total diaria, recibida a la 9 - 14 - 21 h respectivamente. Determinaciones (RIA) de testosterona (T), 17-hidroxiprogestetrona (17-OHP) y cortisol a las 9 - 11 - 14 - 16 - 21 - 23 - 3 - 7 h.
Método estadístico: Anova para medidas múltiples para 2 niveles (programa superanova de Abacus Concepts) y método cosenos para un programa especialmente diseñado (Chronobiology Laboratories. University of Minnesota).
Resultados.T: Presenta ritmo circadiano con cada pauta de tratamiento (p: 0,02). Cada pauta modifica este ritmo (P: 0,0004) (Acrofase con pauta A a las 5h 25 min, acrofase con pauta a las 7h 34 min). Cada tratamiento varía la concentración total de T (p: 0,017), y es menor con la pauta C. 17-OHP: Muestra ritmo circadiano con cada uno de los tratamientos (p: 0,001) y se modifica con cada uno (p: 0,0002) (Acrofase con pauta A: 5h 25 min, acrofase con pauta C: 9h 11 min). La variación de la concentración total de 17-OHP con los distintos tratamientos no tiene significación estadística. Cortisol: Muestra ritmo circadiano con cada tratamiento (p: 0,0025). Los diferentes tratamientos modifican su configuración (p: 0,0001) (Acrofase con pauta A: 11h 36 min; acrofase con pauta B: 23h 36 min; con pauta C: 2 elevaciones: a las 11h y 21 h), correspondiéndose con la toma previa de HC. Las diferentes pautas de tratamiento varían la concentración total de cortisol (p: 0,02) y es mayor con la pauta C.
Conclusiones. El fraccionamiento de la dosis diaria total de HC en pacientes con 21OHD modifica de forma estadísticamente significativa la cronobiología de las hormonas estudiadas. El tratamiento que aporta 2/3 de la cantidad total de HC a las 9 h y 1/3 a las 21 h conduce a un perfil bioquímico mas fisiológico: T, 17-OHP y cortisol mantienen ritmo circadiano similar al normal, es menor la concentración de T y mayor la de cortisol.
Se debe conocer la hora de la extracción de la muestra de control en relación con la toma de HC, por la influencia que ejerce sobre los niveles de hormonas analizadas.
Hiperplasia suprarrenal congénita (HSC) por déficit de 21hidroxilasa: estudio del sistema hla, caracterización genético-molecular y correlación genotipo-fenotipo
J.I. Labarta1, B. Ezquieta2, E. Bello1, L. Marzo3, M.T. Calvo4, E. Mayayo1 y A. Ferrández Longás1
1Unidad de Endocrinología. 3Servicio de Hematología. 4Sección de Genética. Hospital Infantil Universitario Miguel Servet, Zaragoza.
2Becario de Pharmacia. Hospital Infantil La Paz. Madrid.
El objetivo de este trabajo es conocer la utilidad del estudio HLA, caracterizar las mutaciones del gen CYP21 y conocer la correlación genotipo-fenotipo en pacientes afectos de HSC por déficit de 21-hidroxilasa.
Pacientes. Estudio genético de 30 pacientes diagnosticados de HSC [12 pérdida salina (PS), 4 virilizantes simples (VS) y 14 no clásicas (NC)] y de sus familiares (n = 70) procedentes de 26 familias. Estudio del locus A y B del sistema HLA en 25 pacientes no relacionados (12 PS, 3 V S y 10 NC) y sus familiares (n = 62) y comparación con un grupo control (n = 320).
Métodos. Estudio directo del gen (southern blot y reacción en cadena de la polimerasa con hibridación específica de alelo) e indirecto (microsatélites de la región HLA) para despistaje de delecciones y mutaciones puntuales. Serotipado de los locus A y B del HLA.
Resultados.Estudio del gen CYP21. Se consiguió caracterizar el 90,6% de los alelos de las formas clásicas (CL) y el 78,6% de las formas NC. Las mutaciones más frecuentes de los pacientes con formas CL fueron delección (25%) e intrón2G (25%) seguidas de Gln318Stop (21,8%). En las formas NC la más frecuentes fue Val281Leu (32,1%) seguida de Pro453Ser (10,7%) e Ile172Asn (7,1%). No se encontró ninguna mutación de novo. El estudio genético molecular ha permitido identificar 3 pacientes con formas NC crípticas, todos ellos heterozigotos compuestos con la mutación Pro453Ser. 9 de los 14 pacientes (64%) con formas NC eran heterozigotos compuestos para una mutación severa.
Correlación genotipo-fenotipo. Todos los pacientes con afectación enzimática total (grupo null) o casi total (intrón2G) presentaron la forma CL-PS, excepto dos hermanos con forma CL-V S que eran heterozigotos compuestos para intrón2G/conversión parcial (Pro30Leu). Los pacientes con afectación enzimática severa (Ile 172Asn) o parcial (grupo leve ) presentaron una forma V S o NC respectivamente.
Estudio del HLA. La asociación descrita A3 con formas PS no ha sido vista en nuestra serie (20,8% PS vs 22,5% en controles) ni tampoco la asociación B5 con formas VS (4,1% VS vs 14% en controles). Sí se ha observado la asociación B14 con formas NC (32% NC vs 10% en controles, p < 0,05). El 100% de los haplotipos HLA-B14 genotipados han sido Va1281Leu. Se ha observado una asociación negativa (p < 0,05) de los haplotipos Al, A2, B8, B12 y B35 con la HSC. La predicción de la enfermedad realizada en base a los HLA ha sido concordante con el genotipo en todos los casos excepto en una familia. En las familias genotipadas la frecuencia de ser sano, portador o afecto en la descendencia ha sido del 16%, 36% y 48% para las formas CL y del 11,1%, 38,8% y 50% para las NC, respectivamente; estos porcentajes difieren de la esperado para una herencia autosómica recesiva.
Conclusiones. La distribución de mutaciones encontrada en nuestra serie es similar a la comunicada por otros autores a diferencia de una mayor frecuencia de la mutación Gln318Stop del grupo null. Se ha encontrado una buena correlación genotipo-fenotipo. La elevada frecuencia de mutaciones severas en las formas NC hace aconsejable el estudio gen ético en este grupo de pacientes. El estudio HLA en los familiares fue útil para orientar el diagnóstico. Se demuestra una fuerte asociación entre HLA-B14 y la mutación Va1281Leu. Es posible la existencia de una ventaja para transmitir los gametos con mutación del gen CYP21.
Valor predictivo de 17OH progesterona para detección de heterozigosis compuesta con mutación severa en formas no clásicas de deficiencia 21OH (21OHNC) en niños
B. Ezquieta*, A. Oliver, M. Oyarzábal, C. Luzuriaga, R. Gracia, M. Manrique, I. Rica, R. Cano, J. Ramírez, R. Espigares, M. Albisu, Aleixandre, R. Barrio, L. Blasco, M. Borrajo, M. Gutiérrez, J. Labarta, L. Lorenzo y J. Pozo
Bioquímica y Endocrinología Pediátrica. Hospital La Paz. Madrid. Endocrinología Pediátrica y Pediatría del ámbito Nacional.
*Bioquímica del Hospital Gregorio Marañón. Madrid.
La existencia de una fuerte correlación entre severidad clínica y genotipo en las formas clásicas de 21OHD sugiere el interés de valorar si existe tal relación clínico-bioquímica-molecular en formas leves de la deficiencia.
Pacientes y metodos. Se incluyen en este estudio 65 niños con diagnóstico de 21OHNC, establecido a los niveles clínico, bioquímico y molecular. El estudio molecular se ha realizado como se describe en Ezquieta el (Hum Genet 1995, Clin Chem 96, Horm Res 99). El estudio se ha planteado en pacientes completamente genotipados para los que se disponía de los datos cmplementarios aportados por sus Endocrinólogos (incluidos en orden secuencial primario relativo al número de pacientes y alfabético secundario). Se han estudiado comparativamente, en los grupos de pacientes con mutaciones leves versus aquellos heterozigotos compuestos para mutación severa: el nº de signos clínicos y edad al diagnóstico, como indicadores de la severidad clínica; y el metabolito marcador de la deficiencia (17OHP) basal y/o tras estímulo con ACTH, como medida bioquímica del grado de deficiencia. El análisis estadístico se ha realizado mediante t-Student y u-Mann-Witney para variables continuas en pruebas paramétricas y no paramétricas, respectivamente; y chi-cuadrado para discontinuas. El valor predictivo de 17OHP, basal y tras ACTH, para la detección de heterozigosis compuesta con mutación severa se ha analizado mediante curvas ROC.
Resultados y conclusiones. El grado de déficit enzimático, estimado como nivel del metabolito precursor 17OHP, es mayor en pacientes con mutación severa (basal, p = 0,002; tras ACTH p < 0,0001). El análisis mediante curvas ROC ofrece un valor predictivo máximo de la 17OHP-ACTH (area bajo la curva 0,913 DE 0,056) con un 93% de sensibilidad y un 87% especificidad para un punto de corte en 150 nmol/L. Por el contrario, la severidad de la manifestación clínica fue similar en ambos grupos de pacientes (edad p = 0,671; nº signos p = 0,452, al diagnóstico); quedando por valorar la evolución de los mismos. Estos datos ponen de nuevo de manifiesto la fuerte relación déficit enzimático-severidad mutaciones y la disociación clínico-bioquímica también evidente en las formas crípticas de la deficiencia.
El péptido liberador de la hormona de crecimiento (GHRP-6) estimula la expresión de IGF-I en el hipotálamo, cerebelo, e hipocampo de la rata*
L.M. Frago, J. Argente y J.A. Chowen
Hospital Universitario Niño Jesús. Endocrinología y Laboratorio de Investigación. Madrid.
Introducción. Los péptidos liberadores de hormona de crecimiento (GHRPs) son moléculas sintéticas que estimulan la secreción de hormona de crecimiento (GH) desde la hipófisis anterior. En la actualidad, se sabe que estos péptidos actúan sobre los receptores para el nuevo péptido endógeno, ghrelin. Aunque los GHRPs pueden actuar en el hipotálamo para estimular la liberación de GH, su mecanismo de acción aún se ignora. Las acciones de los GHRPs, otras que las de estimular la liberación de GH, aún deben ser determinadas. Los receptores para GHRPs, o ghrelin, se expresan en áreas del sistema nervioso central que no se conoce se encuentren directamente involucradas en la regulación de la GH.
Objetivo. Investigar como afectan los GHRPs al sistema central de IGF.
Diseño experimental y metodología. En ratas Wistar macho adultas se implantó en la vena jugular una minibomba que liberaba 1 µl/hora de solución durante 7 días. Las ratas recibieron suero salino, GHRP-6 (150 µg/día) o GH\100 µg/día). Tras una semana de perfusión, se procedió al sacrificio de los animales, obteniendo sus cerebros. Se disecó el hipotálamo, el cerebelo, el hipocampo y el córtex cerebral, extrayendo el ARN total mediante metodología estándar. Se empleó RT-PCR para determinar los niveles de ARNm de IGF-I.
Resultados. El tratamiento con GHRP-6 indujo un incremento significativo de los niveles de ARNm de IGF-I en el hipotálamo (2,6 veces), cerebelo (1,5 veces) e hipocampo (1,4 veces), pero no en el córtex cerebral. Resultados similares se encontraron en los animales tratados con GH.
Conclusión: Tanto la GH como los GHRPs pueden estimular la expresión de IGF-I en áreas específicas del cerebro, incluyendo aquellas que se desconoce estén involucradas directamente en el control de la secreción de GH.
*Premio Pharmacia a la mejor comunicación oral.
Diferencias auxológicas y hormonales entre pacientes talla baja familiar (TBF) y controles. Análisis génico del receptor de la GH
J.P. López-Siguero*, C.J. del Valle*, L.F. López-Canti*, J.L. Lechuga*, R. Espigares*, M.J. Martínez-Aedo*, R. Cañete*, G. Ródenas*, N. Díaz*, J. Bermúdez*, M. García-Guerrero*, E. Caro*, J.L. Barrionuevo*, A. Gómez-Gila* y A. Ulied**
*Hospitales SAS. **Departamento Médico, Pharmacia.
La TBF ha estado siempre relacionada con factores genéticos y tradicionalmente ha englobado a un grupo heterogéneo de sujetos. En los últimos años se han identificado algunos defectos genéticos que constituirían el fundamento etiológico en un pequeño porcentaje de niños con talla baja (Goddard, 1996).
Objetivos.1) Estudiar parámetros auxológicos y bioquímicos en un grupo de pacientes afectos de TBF (criterios de Ranke) y compararlos con un grupo de sujetos control. 2) Estudiar desde el punto de vista molecular el gen del receptor de la GH en los sujetos con TBF.
Pacientes y métodos. Grupo TBF: 38 pacientes (20 varones y 18 mujeres) con una EC media de 9 ± 2,1 años y una EO de 8,5 ± 2,2 años. Grupo control: 31 sujetos (15 varones y 16 mujeres) con una EC media de 8,4 ± 2,4 años y una EO de 8,2 ± 2,6 años. Los parámetros auxológicos valorados son: talla, velocidad de crecimiento del año previo a la inclusión del estudio (SDS) e IMC; y en el estudio bioquímico: IGF-I, IGFBP-3 expresados en SDS y GHBP expresado en %. El análisis del gen del receptor de la GH se realizó mediante SSCP previa ampliación por PCR del DNA extraído de leucocitos y secuenciación de los fragmentos con patrón de SSCP alterado.
Resultados.

En ambos grupos existen correlaciones positivas significativas entre los niveles de IGF-I y los de IGFBP-3 y GHBP; la IGFBP-3 y la GHBP se correlacionan positivamente con el IMC. Se ha detectado un paciente con patrón alterado en el intrón 6 del gen del receptor de GH, con un doble cambio CA-TG, 30 bases antes del inicio del exón 7.
Conclusiones. Los pacientes con TBF presentan parámetros hormonales (IGF-I, IGFBP-3 y GHBP) significativamente más bajos que los sujetos controles. Los niveles de GHBP se correlacionan con el IMC tanto en los pacientes con TBF como en los controles. Sólo se ha encontrado un paciente con alteración en el gen del receptor de la GH, lo que representa un 2,6% de la población con TBF estudiada.
Este trabajo se ha realizado con una beca Pharmacia.
Diferente neurorregulación de GH en relación a la edad
A. Vela*, R. Coya**, J. Pineda***, M.A. Aniel-Quiroga** y P. Martul*
*Endocrinología Infantil, **Laboratorio de Hormonas, Hospital de Cruces, Barakaldo. ***Departamento de Bioquímica. Universidad del País Vasco (UPV/EHU).
Algunos autores indican que existe un diferente control neuroendocrino de la secreción de GH entre niños y adultos, pero no está sólidamente establecido.
Objetivo. Estudiar la diferente regulación de GH a través de la respuesta de GHRH doble, entre niños prepuberales y adultos y de producirse estudiar la evolución a lo largo de la pubertad para determinar en qué momento tiene lugar.
Material y método. Se seleccionaron 63 niños sanos con talla y velocidad de crecimiento normales y peso adecuado para su talla, edad y sexo. Maduración ósea acorde con la edad cronológica. Talla familiar normal. También 19 adultos jóvenes sanos con talla y peso normales. En ambos grupos se exigió una respuesta de GH superior a 10 ng/ml. Se dividió a los niños en prepuberales, pubertad inicial (estadío II), pubertad media (III+IV) y pubertad tardía (estadío V). El test de GHRH doble consistió en la administración de GHRH (1-29)NH2 a dosis de 1,5 µg/kg i.v. en bolus en los minutos 0 y 120 de la prueba y con muestras cada 15' para determinar GH mediante RIA. Las variables objeto fueron PICO-1 (Respuesta máxima al primer estímulo), PICO-2 (Respuesta máxima tras el segundo estímulo) y proporción de PICO (% de la segunda respuesta respecto de la primera)
Resultados.

Conclusiones.1) Se demuestra el diferente control de la secreción de hormona de crecimiento en adultos con una menor proporción de respuesta respecto de los niños. 2) El hecho de que a lo largo de la pubertad la proporción de pico se mantenga estable indica que este cambio no se produce de forma progresiva sino de forma brusca entre estadío de pubertad tardía y edad adulta.
Estudio de la eficacia de la hormona de crecimiento y análogos de gonadotrofinas en pacientes afectos de talla baja familiar: resultados preliminares al tercer año de seguimiento
L. Castro Feijóo, J. Barreiro Conde, C. Quinteiro García1, J.M. Pérez Gallego2, L. García Alonso2, S. Durán Rivas3, R. Fernández Prieto4, G. González Lorenzo5, D. González Lestón6, A. Buceta Castiñeiras7 y M. Pombo Arias
Unidad de Endocrinología, Crecimiento y Adolescencia. Unidad de Medicina Molecular1. Facultad de Medicina y Complejo Hospitalario Universitario de Santiago de Compostela. Departamento de Pediatría. Hosp. Juan Canalejo2. Centro de Salud de Narón3. Departamento de Pediatría. Hosp. Arquitecto Marcide4.La Coruña. Hosp.Provincial5. Hospital Xeral-Cíes6. Centro de Salud de Cambados7. Pontevedra.
La talla baja idiopática condiciona, sin duda, una talla adulta baja. Ello, junto a la existencia de alteraciones en el eje GH-IGF y el trastorno psicoemocional del paciente, hace que la comunidad científica busque un tratamiento para esta situación de hipocrecimiento.
Objetivos. Determinar la opción terapéutica que consiga el mejor SDS de talla final en niños afectos de talla baja familiar. Establecer cuáles son los parámetros que puedan predecir mejor la respuesta posterior al tratamiento con rhGH y/o análogos GnRH (aGnRH).
Pacientes y métodos. Ensayo clínico abierto, prospectivo, aleatorizado y piloto, randomizado, paralelo con tres grupos de tratamiento. Los sujetos a estudiar son pacientes puberales de ambos sexos con diagnóstico de talla baja familiar. Se incluyeron a pacientes con edad cronológica de 11,5 ± 1 años en niñas y 12,5 ± 1 años en niños (G2 y/o P2 y volumen testicular > 4cc para niños; S2 y/o P2 para niñas). Con velocidad de crecimiento ¾ P50, pronóstico de talla adulta < P10 (Mark2-TW2), SD de talla ¾ P3. Talla materna y/o paterna ¾ P3 (Tanner). Test de secreción de GH: normal. Edad ósea ± 1 año de EC. Edad gestacional: 37-42 sem. Talla y peso al nacer >= P3 (Gaidner-Pearson). Deben dar el consentimiento informado. Los pacientes fueron distribuidos aleatoriamente en tres grupos: Grupo I: control, Grupo II: tratamiento con aGnRH, Grupo III: tratamiento con rhGH + aGnRH. La pauta de tratamiento es: rhGH: 0,9 UI/Kg/semana, análogos de GnRH: 0,06 mg/kg/28 días. Todos los pacientes serán valorados en condiciones basales y cada 3 meses hasta la finalización del crecimiento.
Resultados. La homogeneidad de la muestra, producto de los criterios de inclusión estrictos que se han seguido y que avalan la validez de los resultados, ha ocasionado una gran dificultad en la selección e incorporación de pacientes. Durante el período de inclusión han sido seleccionados 25 pacientes: Grupo I: 9 pacientes. Grupo II: 8 pacientes. Grupo III: 8 pacientes. El análisis estadístico efectuado a los tres años de seguimiento, nos muestra que es el tratamiento con aGnRH + rhGH (III) el que se perfila como la mejor opción terapéutica a seguir en la talla baja familiar, ya que tanto el SDS de la talla como el pronóstico de talla adulta (PTA) mejoran significativamente al compararlos con los grupos I y II. Así como es también este grupo, el que presenta una ganancia mayor en la evolución del SDS de talla y del PTA al compararlos con la talla diana. Además, se aprecia un enlentecimiento de la maduración ósea en los pacientes que reciben tratamiento con aGnRH (Grupos II y III), especialmente en el grupo II, lo que implica una mejoría del PTA de este grupo, pero que, por lo menos de momento, no se acompaña de cambios positivos en cuanto al SDS de la talla. Por último, los datos analizados sugieren que el test de generación de somatomedinas podría ser un predictor de la respuesta al tratamiento con rhGH + aGnRH (Pearson's r = 0,78).
Conclusión. En relación con la respuesta terapéutica y los posibles beneficios sobre la talla final en este tipo de sujetos, los resultados aportados por los diferentes autores resultan conflictivos, en gran medida por tratarse de muestras no homogéneas y sin grupo control. En nuestro estudio, y a pesar de estos resultados preliminares optimistas, será sólo al final del mismo cuando se pueda llegar a conclusiones en cuanto a la efectividad del tratamiento de la talla baja familiar, así como a la identificación de los parámetros que permitan seleccionar a los pacientes susceptibles de una mejor respuesta terapéutica.
Trabajo realizado con ayuda del FIS y los laboratorios Pharmacia &Upjohn e Ipsen Pharma.
Estudio longitudinal del brote de crecimiento puberal en los niños nacidos pequeños para la edad de gestación (SGA) sin recuperación postnatal
E. Vicens-Calvet*, A. Carrascosa* y R.M. Espadero**, en nombre del Grupo Colaborativo Español para el Estudio del Retraso
de Crecimiento Intrauterino
*Hospital Universitario Materno-Infantil Vall D'Hebron. **Pharmacia SA.
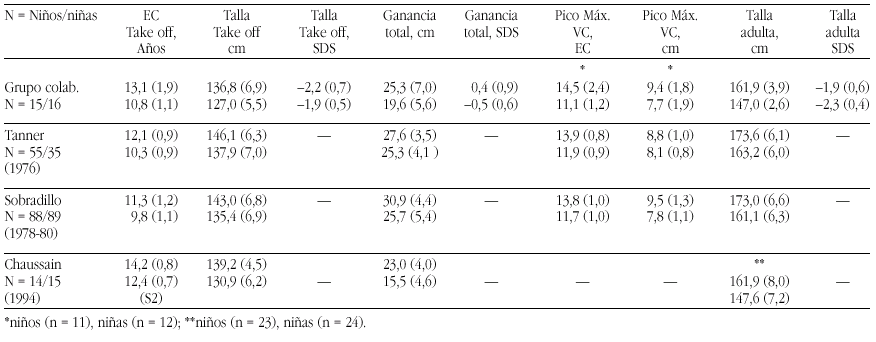
Objetivos. Realizar un estudio longitudinal del brote de crecimiento puberal (BCP) en un grupo de niños con SGA sin recuperación postnatal de talla, para determinar sus características y posible repercusión sobre la talla baja adulta que presentan estos pacientes.
Material. De una cohorte total de 553 SGA diagnosticados según las tablas de Gairdner & Pearson y entre 2-8 años talla < P3 según Tanner Whitehouse, se estudió longitudinalmente en 31 casos (15 niños y 16 niñas) las principales características del BPC.
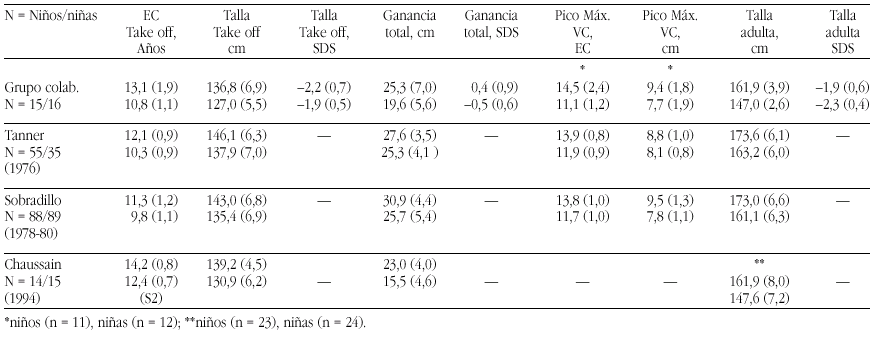
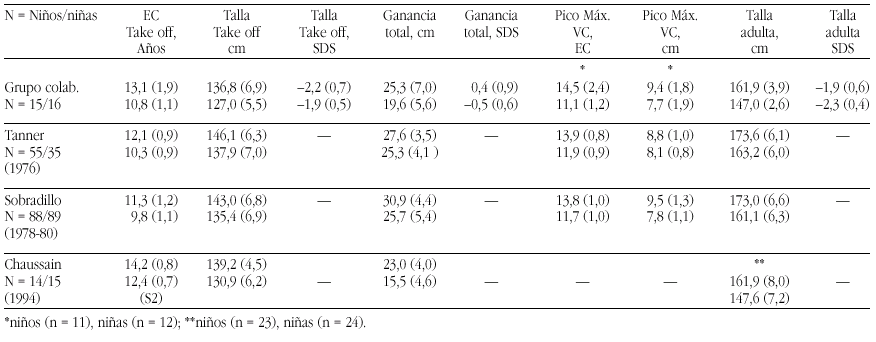
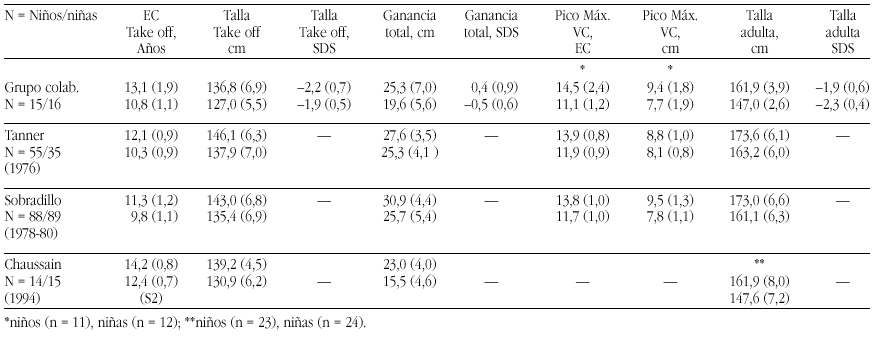
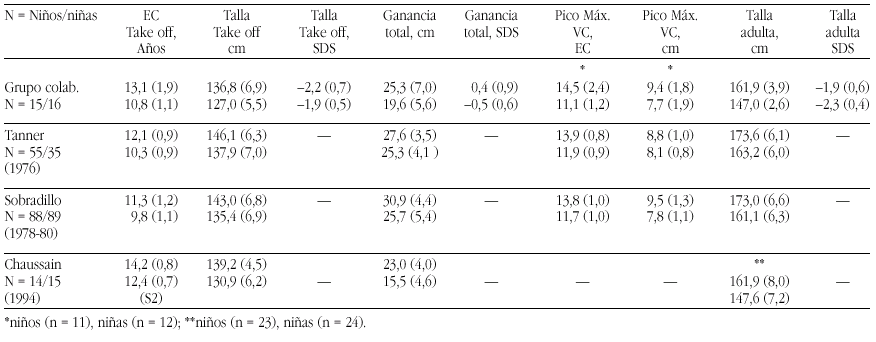
Métodos. Las curvas de itinerario durante la pubertad hasta la talla final fueron analizadas según un programa KGS 4.5 para determinar la Edad Cronológica (EC) al inicio del despegue puberal ("take-off"), ganancia puberal total, pico de máxima velocidad de crecimiento (PMV), talla final (TF) absoluta y TF SDS. Se han comparado los resultados de nuestro grupo con los de población estándar (Tanner et al., Sobradillo et al.) y con con el estudio de Chaussain et al.
Resultados. Véase tabla a pie de página.
Comentarios.Niños: El "take off" se inicia más tarde que en las poblaciones de Tanner (p < 0,05) y Sobradillo (p < 0,0005). La ganancia total es similar a la del grupo de Tanner (p < 0,2) e inferior a la de Sobradillo (p < 0,005) y el PMV es normal. El BCP es muy aceptable y supone una ganancia de 0,4 SD. Niñas: El "take off" se inicia a la misma edad que en la población de Tanner (p < 0,1) y significativamente más tarde al estudio de Sobradillo (p < 0,0005). La ganancia total es inferior tanto a la de Tanner (p < 0,0005) y Sobradillo (p < 0,005) y el PMV es normal. El BCP inferior a la normalidad supone una pérdida de 0,5 SD. Es difícil la comparación con el grupo de Chaussain ya que todos sus datos parten de S2/G2.
Grupo colaborativo:Alpera R, Álvarez E, Bel J, Borrajo E, Borrás V, Cabrinety N, Caimari M, Carrascosa A, Castro R, Closas R, Costa I, Chueca M, Del Valle CJ, Espigares RM, Ferragut J, Ferrández Arenas A, Ferrández Longás A, González Díaz JP, Gracia Bouthelier R, Gutiérrez P, Hermoso F, Hollenberg G, López García MJ, López Siguero JP, Lorenzo L, Luzuriaga C, Marcos V, Martínez Aedo MJ, Martul P, Nieto JA, Oliver A, Oyarzábal M, Pombo M, Prieto J, Rica I, Rivas Crespo MF, Rodríguez Hierro F, Rodríguez I, Roldán B, Sobradillo B, Tacons J, Vargas F, Vicens-Calvet E, Yeste D, Yturriaga R, Ulied A, Espadero RM.
Complicaciones endocrinológicas en pacientes sometidos a trasplante de progenitores hematopoyéticos en la edad pediátrica
G. Carreras*, M. Hernández**, E. López*, I. Badell*, N. Pardo*,
A. Pérez** y J. Cubells*
Servicios de *Pediatría y **Endocrinología. Hospital Sant Pau. UAB. Barcelona.
La mayor experiencia en trasplantes de progenitores hematopoyéticos (TPH) y la mayor supervivencia de los pacientes sometidos a estas técnicas han conducido a que cada vez se de más importancia a las alteraciones endocrinológicas derivadas de estos tratamientos.
Objetivo. Valorar el tipo y la prevalencia de las alteraciones endocrinológicas en los pacientes sometidos a TPH en edad pediátrica en nuestro hospital.
Pacientes y métodos. Entre octubre 1984 y enero 1998 se practicaron 271 TPH en 271 pacientes. Estudiamos 69 pacientes (54% varones, 93% afectos de enfermedades malignas) sometidos a 72 TPH, que sobreviven libres de enfermedad y con seguimiento posterior en nuestro centro. El 26% de los TPH fueron alotrasplantes y en el 61% se practicó irradiación corporal total (ICT) como parte del tratamiento previo. La edad media en el momento del trasplante fue de 8,2 ± 4,2 años (0-16), y el seguimiento medio de 7,2 ± 2,6 años. El 83% de los pacientes estaban en un estadio I de Tanner y 5 habían alcanzado la talla definitiva al realizarse el TPH. En 50 pacientes se realizaron determinaciones seriadas de hormonas tiroideas, y de gonadotrofinas en 37 de los 55 pacientes que habían alcanzado la edad puberal. En todos se valoraron el peso y la talla en cada visita.
Resultados. Se detectó un hipotiroidismo primario en el 52% de los pacientes, siendo transitorio en 7/26. En el 78% (14/19 mujeres; 15/18 hombres) de los pacientes que habían alcanzado la edad puberal se evidenció un hipogonadismo hipergonadotrófico. En el 100% de las mujeres existía aumento tanto de la LH como de la FSH, mientras que en los hombres la afectación era casi exclusiva de la línea germinal. Los 3 varones que mostraron elevaciones de los niveles de LH, tenían niveles de testosterona séricos normales y presentaron una virilización espontánea. En cuanto a la talla, eliminando los 5 pacientes con talla inicial definitiva, se observó una pérdida media de 0,56 desviaciones estándar (ds) al final del seguimiento, siendo de 0,91 ds y 0,66 ds en los que tenían niveles de LH elevados o normales, respectivamente. No se observaron diferencias entre sexos, tipo de trasplante, estadio de Tanner inicial ni final, ni tratamiento con ICT.
Conclusión. Dadas la elevada prevalencia y la relevancia de las alteraciones endocrinológicas asociadas al tratamiento con TPH, es necesario realizar el despistaje sistemático de las mismas, así como su tratamiento precoz y adecuado.

