

El tumor miofibroblástico inflamatorio (TMI) es una lesión infrecuente de malignidad intermedia que aparece en las primeras décadas de la vida1,2. Descrita en múltiples localizaciones, el pulmón es el lugar más afectado1, seguido del abdomen y, en menor frecuencia, cerebro, órbita, partes blandas y tracto genitourinario2,3. La etiología es desconocida, aunque se han descrito algunos factores predisponentes como cirugías, traumatismos de repetición, infecciones crónicas o radioterapia previa2,3.
La clínica varía según la localización. Son frecuentes los síntomas compresivos locales, pudiendo producir tos, hemoptisis, dolor torácico o atelectasias en los tumores de localización pulmonar, así como dolor abdominal o melenas en los de origen abdominal1. Es asintomático en hasta un 40%, detectándose de manera casual en pruebas de imagen1. Con frecuencia se acompaña de anemia, trombocitosis, leucocitosis, elevación de velocidad de sedimentación globular (VSG) e hipergammaglobulinemia, sobre todo en los de origen abdominal1. Los hallazgos por imagen son inespecíficos, típicamente se identifica una lesión sólida bien definida, irregular, que puede presentar calcificaciones hasta en un 25% de los casos2. En estudios con contraste puede mostrar realce heterogéneo en fase tardía2. El diagnóstico requiere descartar otro tipo de tumores, abscesos e infecciones crónicas (aspergilosis o micobacterias)1. La confirmación diagnóstica se obtiene con la histología, la cual muestra miofibroblastos en forma de huso junto con cantidad variable de células inflamatorias (eosinófilos, células plasmáticas y linfocitos)2,3, siendo típicamente positivos para vimentina, actina, CD34 y CD1172,3. La reactividad a quinasa del linfoma anaplásico (ALK) se da en un 50% de los pacientes, más frecuente en jóvenes y asociada a recurrencias locales, pero no a metástasis a distancia, las cuales se limitan, habitualmente, a lesiones ALK negativas4.
El tratamiento de elección es la exéresis completa del tumor, curativa en la mayoría de los casos2. Sin embargo, es necesario un seguimiento estrecho ya que, en algunos casos, el tumor puede recidivar o malignizar, siendo necesario asociar tratamientos adyuvantes como quimioterapia, radioterapia o inmunosupresores1. En aquellos tumores con positividad para ALK se dispone de herramientas terapéuticas específicas: los inhibidores de ALK5,6. Estos fármacos juegan un papel importante ya que pueden utilizarse en casos de difícil resección como neoadyuvantes a la cirugía o en recidivas locales y/o metastásicas, logrando un aumento global de la supervivencia5. El crizotinib fue uno de los primeros fármacos en utilizarse, sin embargo, están descritos casos de mala respuesta inicial o de adquisición de resistencias tras meses de tratamiento, lo que ha motivado el desarrollo de inhibidores de ALK de «segunda generación» (ceritinib, alectinib) con resultados prometedores6.
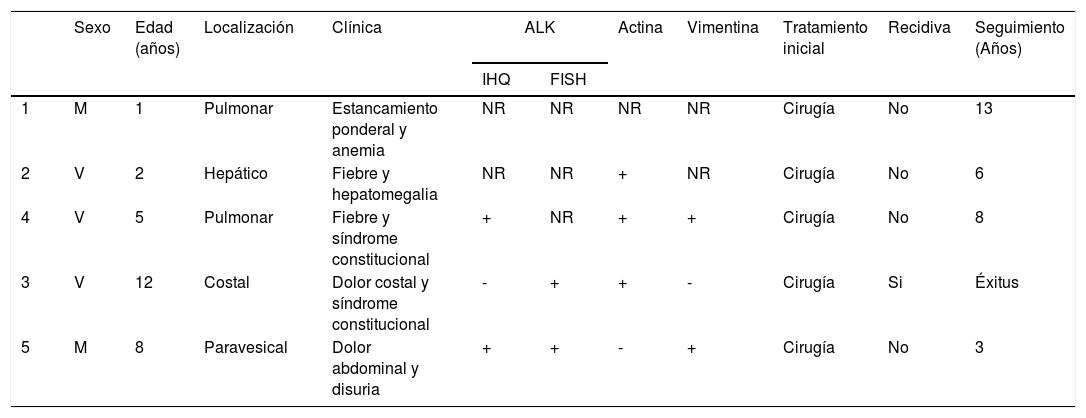
Con el objetivo de conocer la casuística en un centro terciario, se ha realizado un estudio descriptivo retrospectivo de pacientes menores de 16 años, entre los años 2005 y 2020, encontrándose cinco casos diagnosticados. Se han analizado las siguientes variables: edad al diagnóstico, sexo, clínica al debut, localización del tumor, exploraciones complementarias realizadas, tratamiento y evolución (tabla 1).
Serie de casos de tumor miofibroblástico inflamatorio revisados en nuestro centro
| Sexo | Edad (años) | Localización | Clínica | ALK | Actina | Vimentina | Tratamiento inicial | Recidiva | Seguimiento (Años) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IHQ | FISH | ||||||||||
| 1 | M | 1 | Pulmonar | Estancamiento ponderal y anemia | NR | NR | NR | NR | Cirugía | No | 13 |
| 2 | V | 2 | Hepático | Fiebre y hepatomegalia | NR | NR | + | NR | Cirugía | No | 6 |
| 4 | V | 5 | Pulmonar | Fiebre y síndrome constitucional | + | NR | + | + | Cirugía | No | 8 |
| 3 | V | 12 | Costal | Dolor costal y síndrome constitucional | - | + | + | - | Cirugía | Si | Éxitus |
| 5 | M | 8 | Paravesical | Dolor abdominal y disuria | + | + | - | + | Cirugía | No | 3 |
FISH: hibridación fluorescente in situ; IHQ: inmunohistoquímica; M: mujer; NR: no realizado; V: varón.
Se recogieron un total de cinco casos, tres de ellos varones y dos mujeres. La edad media al diagnóstico fue de 5,6 años (rango de 1,8 a 12,2 años). Encontramos dos tumores de localización pulmonar, dos abdominales y uno óseo. En la serie recogida, la clínica de debut fue principalmente debido al efecto masa del tumor (dolor costal y dolor abdominal) pero todos los pacientes asociaban síndrome constitucional. Un paciente se diagnosticó inicialmente de tuberculosis pulmonar y otro de absceso hepático. A nivel analítico, destacó la presencia de trombocitosis (cuatro casos), anemia (tres), elevación de proteína C reactiva (tres) VSG (dos) y lactato deshidrogenasa (dos).
Tras una primera valoración radiológica (fig. 1), el diagnóstico fue confirmado con biopsia en los cinco casos, completándose la anatomía patológica con estudio inmunohistoquímico y hibridación fluorescente in situ en los casos más recientes (ALK + en 3/3, actina + en 3/4 y vimentina + en 2/3). El tratamiento fue la exéresis del tumor en todos los casos con buena evolución en 4/5 niños a lo largo de una mediana de seguimiento de siete años. Un paciente presentó malignización del tumor (fibrosarcoma inflamatorio) y recaída precoz tras cirugía, requiriendo tratamiento adyuvante con ceritinib con una rápida respuesta inicialmente pero posterior progresión a los meses y éxitus.
 Radiografía de parrilla costal muestra infiltración y destrucción parcial del arco posterior de la octava costilla izquierda (flecha blanca), asociando una gran masa de partes blandas intratorácica extrapulmonar con tenues calcificaciones amorfas (*) y discreto derrame pleural acompañante. B) Ecografía de la masa intratorácica heterogénea. C) Resonancia magnética secuencia T2 coronal y D) T1 transversal con saturación grasa y contraste intravenoso, que muestran una masa lobulada en contacto con la pleura mediastínica, pericardio, pleura axilar y diafragmática y realce periférico (punta de flecha) con área de necrosis central.")
A) Radiografía de parrilla costal muestra infiltración y destrucción parcial del arco posterior de la octava costilla izquierda (flecha blanca), asociando una gran masa de partes blandas intratorácica extrapulmonar con tenues calcificaciones amorfas (*) y discreto derrame pleural acompañante. B) Ecografía de la masa intratorácica heterogénea. C) Resonancia magnética secuencia T2 coronal y D) T1 transversal con saturación grasa y contraste intravenoso, que muestran una masa lobulada en contacto con la pleura mediastínica, pericardio, pleura axilar y diafragmática y realce periférico (punta de flecha) con área de necrosis central.
El TMI es un tumor de malignidad intermedia. La clínica está determinada por el sitio anatómico afectado y los signos inflamatorios sistémicos. El manejo dependerá de la localización, factibilidad de la resección quirúrgica, comportamiento y expresión de ALK. El tratamiento con inhibidores de ALK parece tener resultados alentadores. Los hallazgos descritos en nuestra serie son similares a los encontrados en la bibliografía revisada. Se aporta a la literatura un tumor de localización costal, menos frecuentemente descrito.
