



El síndrome de neoplasia endocrina múltiple tipo 2B (MEN2B) es la forma menos frecuente pero más agresiva de MEN2 por el precoz desarrollo de carcinoma medular de tiroides (CMT), presente en todos los casos. Asocia feocromocitoma y un fenotipo muy característico1,2. Está producido por una mutación activadora del protooncogén RET de herencia autosómica dominante, aunque en > 50% no tiene asociación familiar2. El 95% de los pacientes presenta la mutación M918T3. En relación con el genotipo, la Asociación Americana de Tiroides clasifica 4 grupos de riesgo para desarrollar neoplasias que determinarán el seguimiento y el tratamiento profiláctico1. En los pacientes con riesgo elevado, que son la mayoría por estar M918T incluida, se recomienda cirugía profiláctica antes del año de edad1,4.
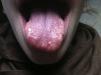
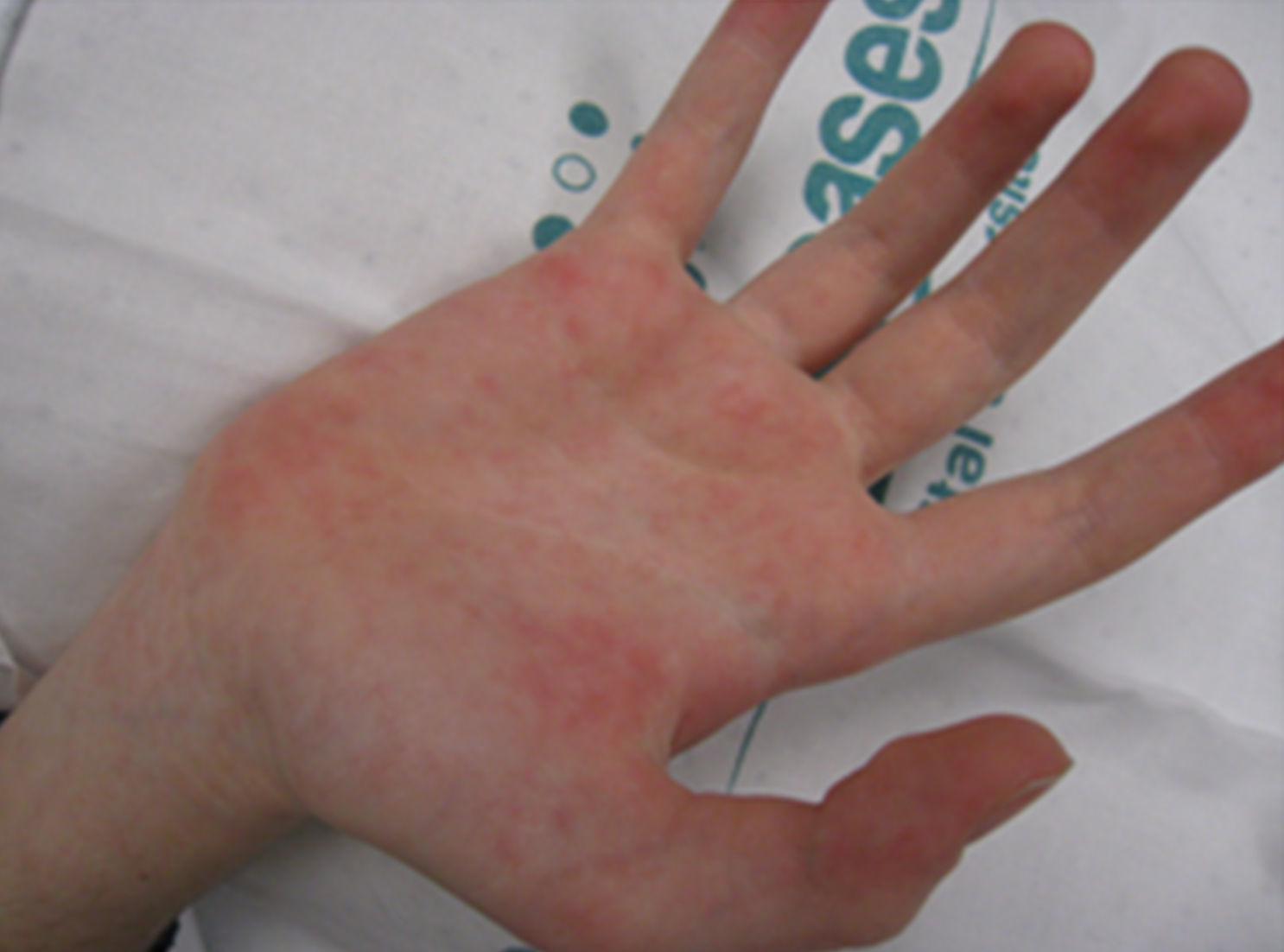
Presentamos a una niña de 13 años con un bocio multinodular detectado 6 meses antes, función tiroidea normal y una ecografía compatible. Antecedentes: dismetría de extremidades y alteración de los vasos retinianos. Presenta hábito marfanoide, labios gruesos, nódulos linguales (fig. 1) y un bocio visible de consistencia pétrea. Antropometría: 162cm (+0,67 SDS), 42 kg (–1 SDS) e IMC 16kg/m2(–1,26 SDS). Destacan niveles de calcitonina de 9.581 ng/ml, con PTH y catecolaminas urinarias normales. La ecografía con punción-aspiración con aguja fina (PAAF) evidencia 3 nódulos con microcalcificaciones y un patrón vascular mixto (uno en el lóbulo tiroideo izquierdo [2,4×2,7×3,9] y dos en 3l lóbulo tiroideo derecho [1,2×0,8×1 y 1,7×1,2×2,6]). La PAAF confirma el CMT y la TC, nódulos tiroideos con probable extensión local. El estudio del protooncogén RET confirma la mutación M918T, con estudio familiar negativo. Se realiza una tiroidectomía total y vaciamiento ganglionar bilateral. Se objetiva extensión local tumoral a recurrente derecho, pared de tráquea y esófago, que son reseccionados parcialmente en un segundo tiempo quirúrgico, quedando mucosa esofágica infiltrada residual. Postoperatoriamente, se inicia tratamiento con L-tiroxina, así como calcitriol y calcio por hipocalcemia. Tres semanas tras la cirugía, al tratarse de una resección incompleta y teniendo en cuenta un caso descrito de similares características con buena respuesta a sorafenib5, se decide iniciar tratamiento paliativo a 10mg/kg/día cada 12 h. En controles posteriores, la calcitonina desciende hasta 133 ng/ml y las ecografías no presentan signos de recidiva. Durante el tratamiento con sorafenib asocia pérdida ponderal, diarrea y síndrome mano-pie (fig. 2) por lo que, tras 8 meses, se suspende.
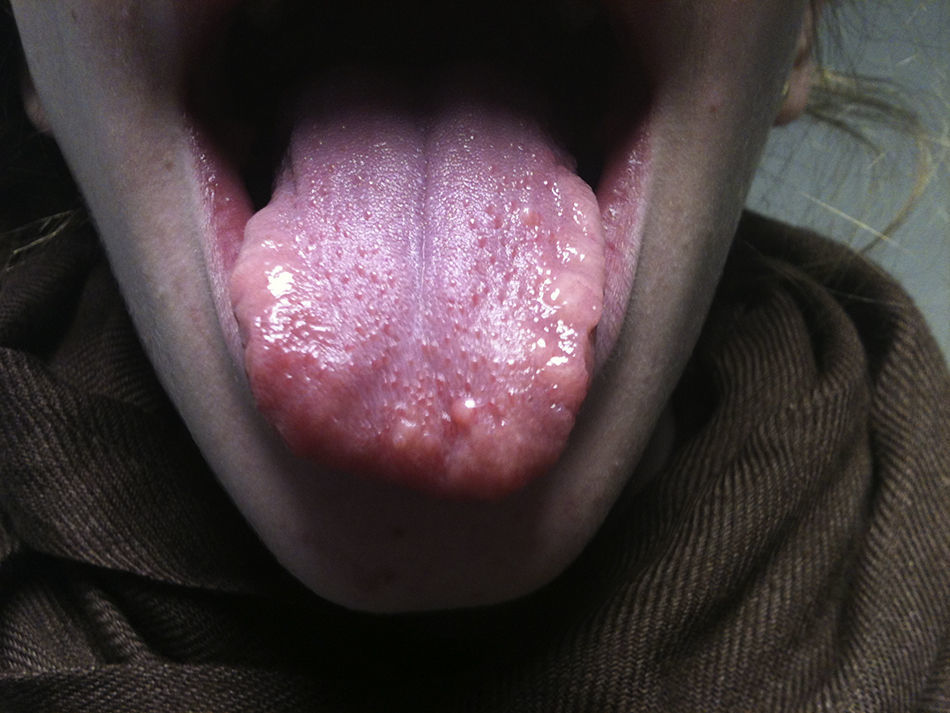
El reconocimiento clínico será la única forma de pensar en MEN2B esporádico. Debemos sospecharlo ante todo paciente con neuromas, principalmente si asocia hábito marfanoide (cara alargada, extremidades alongadas y manos y pies grandes). Los neuromas mucosos afectan a todo el tracto gastrointestinal, especialmente boca, lengua y labios, y acostumbran a aparecer a partir de los 2 años. Antes pueden presentar alteraciones inespecíficas del hábito intestinal. También asocian anomalías esqueléticas, sequedad ocular o conjuntivitis, etc.1,2,6. Aunque se ha descrito el CMT en menores de 6 meses, se diagnostica frecuentemente por una tumoración cervical a edades mayores y en un estadio avanzado1. El 50% de los pacientes asocian feocromocitoma, que frecuentemente aparece entre la 2a y la 3a décadas, es intraadrenal y de curso benigno1.
Ante la sospecha de MEN2B, se deberá solicitar un estudio que incluya calcio iónico, calcitonina y antígeno carcinoembrionario (CEA). Se estudiará el protooncogén RET y se realizará una ecografía de cuello. Si se evidencia tumoración, se practicará una PAAF para descartar CMT. En los mayores de 8 años con MEN2B, se deberán realizar controles anuales de catecolaminas en sangre/orina, que se completarán con ecografía abdominal si hay sospecha clínica o bioquímica1,2.
El CMT determinará el pronóstico1,2,4. Presenta niveles de CEA y calcitonina elevados, siendo el segundo más específico y de mayor valor pronóstico7. El diagnóstico definitivo es histológico y es preciso realizar estudios de extensión1,2. Es resistente a quimioterapia y radioterapia, y el único tratamiento curativo es la resección completa1,2,7-9. Si se evidencia enfermedad metastásica, estará indicado el tratamiento paliativo1,2. Destacan los inhibidores de la tirosincinasa porque, a pesar de estar en fase de estudio, sí parecen inducir una respuesta, al menos parcial2,5,7-9. Asocian efectos adversos como hipertensión, diarrea, pérdida de peso o reacción cutánea mano-pie5,7-9. Dado que muchos pacientes pueden estar asintomáticos largo tiempo y que no existe evidencia de mayor respuesta si se inician precozmente, se recomienda posponerlos hasta progresión de la enfermedad9.
Presentamos a una paciente que desde la primera infancia presenta rasgos compatibles con MEN2B y que al diagnóstico ya presentaba un CMT en estadio avanzado. Este caso ilustra la necesidad de los pediatras de conocer el fenotipo propio del síndrome para poder hacer un diagnóstico precoz y un tratamiento profiláctico.
