




Los errores innatos del metabolismo intermediario (EIMi) son enfermedades genéticas heterogéneas que causan importante morbimortalidad y representan un reto diagnóstico. El objetivo de este trabajo es describir el número, el tipo y las características clínicas de los pacientes con EIMi en un hospital pediátrico de alta especialidad.
Material y métodosEstudio retrospectivo de 204 expedientes de pacientes diagnosticados con EIMi por sospecha clínica, de enero del 2000 a diciembre del 2012, analizados antes y después de la implementación de la espectrometría de masas en tándem (MS/MS) como herramienta de tamiz selectivo.
ResultadosEn los 204 casos analizados, se encontraron 25 diferentes tipos de EIMi: 102 con acidurias orgánicas y 100 con aminoacidopatías y 2 con defectos de la beta oxidación. La introducción de la MS/MS incrementó el número de casos detectados en 50%. Los pacientes fueron enviados por 13 diferentes servicios médicos, siendo los pediatras los que remitieron más casos. El intervalo promedio entre el inicio de los síntomas y el diagnóstico fue de 18 meses.
ConclusiónEn los niños enfermos mexicanos estudiados se encontró una gran variedad de EIMi, destacando los defectos del propionato y la enfermedad de orina de jarabe de arce. En esta población analizada, el diagnóstico de la enfermedad metabólica se realizó en forma muy tardía. Estos resultados pueden servir como evidencia para incorporar los EIMi al tamiz neonatal ampliado, o en su defecto para que se realice el diagnóstico selectivo en todos los niños hospitalizados con datos clínicos indicativos.
Inborn errors of intermediary metabolism (IEiM) are a group of heterogeneous genetic diseases that are diagnostically challenging and cause significant morbidity and mortality. The aim of this study is to perform a descriptive analysis of the number, type, and clinical features, in a series of cases with IEiM identified through selective diagnosis in a highly specialized pediatric hospital.
Materials and methodsA retrospective study was performed from January of 2000 to December of 2012 by analyzing the files of 204 patients with an IEiM, by selective screening, before and after the implementation of tandem mass spectrometry (MS/MS).
ResultsA total of 25 different types of IEiM were found in the 204 files; 102 organic acidurias, 100 aminoacidopathies, and 2 fatty acid oxidation disorders. The introduction of MS/MS increased the number of cases detected by 50%. Patients were referred from 13 different specialists, with pediatricians being the most active. The average interval between onset of symptoms and diagnosis was 18 months.
ConclusionAmong the sick Mexican children studied, a wide variety of IEiM was found, propionate defects and maple syrup urine disease being noteworthy. The diagnosis of metabolic disease was delayed in the population studied. These results present evidence to perhaps incorporate IEiM into an expanded newborn screening, or else to perform selective diagnosis in all hospitalized children with suggestive clinical data.
Los errores innatos del metabolismo (EIM) son un grupo complejo y heterogéneo de trastornos monogénicos cuyas consecuencias clínicas son generalmente graves y causan importante morbimortalidad, primordialmente en los pacientes pediátricos1-4. Dentro de ellos, los EIM intermediario (EIMi) son aquellos en los que el defecto genético afecta una enzima localizada en alguna de las vías metabólicas responsables de transformar las proteínas, hidratos de carbono y lípidos en equivalentes reducidos que al ser introducidos en el sistema de fosforilación oxidativa mitocondrial producen ATP, que es la unidad energética que todas las células del organismo necesitan5.
Desde el punto de vista fisiopatológico, la mayoría de los EIMi entran en las categorías de defectos de tipo intoxicación o energético (grupos 1 y 2 de la clasificación propuesta por Saudubray)6 y predominantemente presentan en su curso evolutivo problemas agudos que requieren atención urgente, especialmente en el periodo neonatal y del lactante, aunque ocurren en cualquier otra época de la vida. Dentro de sus características clínicas más notables, destacan las manifestaciones neurológicas y digestivas, pero pueden afectar a todos los órganos y sistemas del cuerpo2.
En las últimas décadas el escenario de estas enfermedades ha cambiado gracias a las nuevas metodologías de diagnóstico, tales como la espectrometría de masas en tándem (MS/MS), que permiten el reconocimiento presintomático de sus biomarcadores en etapas tempranas de la vida, y a los mejores tratamientos médicos7.
Se estima que de manera colectiva, los EIM afectan entre 1:500 a 1:1,500 recién nacidos1,8,9 y la presencia de estos trastornos entre los niños enfermos es aún mayor10,11. Generalmente, los datos consistentes sobre la frecuencia de los EIM provienen de la información generada por los sistemas de tamiz (cribado) neonatal ampliado (TNA) y se sabe que existen diferencias en la prevalencia de los EIM dependiendo de la etnicidad de la población analizada12,13.
En México, estas afecciones han sido poco estudiados, no existen registros nacionales, su detección mediante el TNA no es obligatoria y en general, se desconoce su frecuencia, tanto en la población de recién nacidos vivos como entre los niños enfermos. El objetivo de este trabajo es dar a conocer el número, el tipo y las características clínicas de los pacientes con EIMi en un hospital pediátrico de alta especialidad, mediante el diagnóstico selectivo.
Material y métodosSe realizó un estudio retrospectivo de 204 casos diagnosticados con algún EIMi de enero del 2000 a diciembre del 2012 en el Instituto Nacional de Pediatría (INP), que es un centro gubernamental nacional de tercer nivel de atención en la ciudad de México y que recibe pacientes de instituciones médicas de todo el país. En función de las técnicas y los recursos disponibles en el momento del estudio, solamente fueron considerados los defectos de aminoácidos, trastornos de la beta oxidación de los ácidos grasos y acidurias orgánicas. Se documentaron el número y el tipo de EIMi, el cuadro clínico y los principales datos demográficos. La plataforma analítica incluyó cromatografía de líquidos de alta resolución (HPLC), cromatografía de gases acoplada a espectrometría de masas (GC/MS) y espectrometría de masas en tándem (MS/MS); Se analizó el número de pacientes diagnosticados en 2 etapas: a) aquellos detectados con HPLC y GC/MS, y b) los detectados después de la implementación de la MS/MS a partir de enero del 2010. Dependiendo del caso, se utilizaron otras pruebas complementarias tales como cuantificación de ácido orótico. Se calculó la correlación entre la edad de inicio de los síntomas referidos por los padres o los médicos y la edad al momento del diagnóstico bioquímico definitivo emitido por el Laboratorio de Errores Innatos del Metabolismo y Tamiz del Instituto Nacional de Pediatría, para cada paciente clasificado por grupo etario.
ResultadosDe los 204 casos documentados con EIMi, 97 fueron mujeres y 107 varones, provenientes de 192 familias; 12 familias (6,2%) tuvieron 2 hijos afectados. Se registró consanguinidad en 26/192 familias (13,5%). Treinta y nueve (20,3%) familias tuvieron el antecedente de hijos con muertes inexplicables en la infancia y 47 (24,5%) contaron con el antecedente de otros hijos afectados con un cuadro clínico similar al del propósito. El 77,5% de los niños fueron de término, el 18% pretérmino y el 4,5% postérmino. Los pacientes provinieron de 29 de los 32 estados que conforman el país.
Se encontraron 25 diferentes tipos de EIMi: 102 pacientes con acidurias orgánicas, 100 con aminoacidopatías y 2 con defectos de la beta oxidación de los ácidos grasos (tabla 1). En la tabla 2 se observa que la mayor proporción de pacientes correspondieron a lactantes menores y que el número de casos diagnosticados aumentó de 14 a 21 por año desde que se incorporó el equipo de MS/MS a la plataforma analítica de nuestro laboratorio (etapa ii). Los pacientes fueron enviados por 13 tipos distintos de servicios médicos, siendo los pediatras generales los que remitieron un mayor número de ellos (21,6%), seguidos por los especialistas en urgencias y terapia intensiva (18,1%); genética (16,6%); neonatología (14,7%) y neurología (13,3%). El resto de los pacientes (15,8%) fue enviado por otros especialistas (gastroenterólogos, infectólogos, nefrólogos, psiquiatras, oftalmólogos, endocrinólogos y hematólogos). También se documentaron 2 casos de PKU que fueron enviados por el padre de una paciente con dicha enfermedad.
EIMi diagnosticados en el Instituto Nacional de Pediatría de México del año 2000 al 2012, agrupados por categorías
| TipoEIMi | Enfermedad | Abreviatura | Número de casos n=204 pacientes |
| Acidurias orgánicasn=102 | Acidemia metilmalónica | MMA | 54 |
| Acidemia propiónica | PA | 15 | |
| Acidemia isovalérica | IVA | 6 | |
| Deficiencia de β-cetotiolasa | BKT | 4 | |
| Acidemia láctica primaria | LA | 4 | |
| Aciduria glutárica tipo i | GA-I | 4 | |
| Aciduria 3-hidroxi-3metilglutárica | HMG | 4 | |
| Deficiencia múltiple de carboxilasas | MCD | 3 | |
| Deficiencia de 2-metil 3-hidroxi-butiril-CoA deshidrogenasa | 2M3HBA | 3 | |
| Deficiencia de succinil-CoA acetoacetato transferasa | SCOT | 2 | |
| Deficiencia de aspartoacilasa | ASPA | 2 | |
| Encefalopatía etilmalónica | EE | 1 | |
| Trastornos de los aminoácidosn=100 | Enfermedad de orina de jarabe de arce | MSUD | 30 |
| Hiperfenilalaninemias/fenilcetonuria | H-PHE/PKU | 23 | |
| Tirosinemia tipo i | TYR-I | 10 | |
| Citrulinemia | CIT-I | 9 | |
| Deficiencia de ornitina transcarbamilasa | OTC | 8 | |
| Homocistinuria | HCY | 6 | |
| Deficiencia de argininosuccinil-CoA liasa | ASA | 3 | |
| Argininemia | ARG | 6 | |
| Defectos de la tetrahidrobiopterina | BIOPT | 3 | |
| Hiperglicinemia no cetósica | NKHG | 1 | |
| Atrofia girata | OAT | 1 | |
| β-oxidaciónn=2 | Deficiencia de 3-hidroxi-acil-CoA deshidrogenasa de cadena larga | LCHAD | 1 |
| Deficiencia de acil-CoA deshidrogenasa de cadena media | MCAD | 1 |
Las abreviaturas se señalan según el American College of Medical Genetics14.
Número de pacientes diagnosticados según su grupo de edad, por método analítico utilizado antes (etapa i) y después de la incorporación de la MS/MS (etapa ii)
| Grupo de edad | Número de casos | Etapa I HPLC/CGEM en 10 años | Etapa II HPLC/CGEM+MS/MS en 3 años |
| Recién nacidos(0-30 días) | 23 | 15 | 8 |
| Lactantes menores(1-23 meses) | 123 | 86 | 37 |
| Lactantes mayores(24-36 meses) | 22 | 17 | 5 |
| Preescolares(3-6 años) | 14 | 8 | 6 |
| Escolares y adolescentes(> 6 años) | 22 | 14 | 8 |
| Casos diagnosticados por año | 15.7 | 14 | 21 |
| Total | 204 | 140 | 64 |
Los principales datos clínicos que motivaron la sospecha diagnóstica se muestran en la tabla 3, desglosados por aparatos y sistemas, destacando los hallazgos bioquímicos y los signos y síntomas neurológicos como los más frecuentes.
Manifestaciones clínicas de los 204 pacientes presentes al momento del diagnóstico agrupadas por aparatos y sistemas
| SISTEMA NERVIOSO CENTRAL | HALLAZGOS BIOQUÍMICOS | SISTEMA HEMATOPOYÉTICO |
| Retraso psicomotor (115)Crisis convulsivas (102)Hipotonía (101)Irritabilidad (83)Deterioro neurológico (75)Succión débil (66)Letargo (61)Somnolencia (51)Encefalopatía (41)Espasticidad (29)Trastorno del comportamiento (26)Estupor (22)Retraso del lenguaje (20)Retraso mental (17)Ataxia (13)Coma (13)Distonía (9)Edema cerebral (8)Tremor (7)Síndrome demencial (5)Neuroinfección (4)Hemorragia cerebral (3)Infarto cerebral (3) | Acidosis metabólica (115)Hiperamonemia (70)Anión gap aumentado (51)Cetosis (43)Deshidratación (37)Hipoglucemia (26)Acidemia láctica (24)Alcalosis (7)APARATO DIGESTIVORechazo al alimento (80)Vómito (72)Desnutrición (48)Hepatomegalia (27)Ictericia (26)Enfermedad por reflujo gastroesofágico (20)Diarrea (19)Insuficiencia hepática (11)Esplenomegalia (9)Cirrosis (4)APARATO RESPIRATORIODificultad respiratoria (48)Crisis de apnea (48)Cianosis (25)Taquipnea (18)Neumonía (16)Paro cardiorrespiratorio (9) | Trombocitopenia (31)Tiempos de coagulación prolongados (13)Anomalías tromboembólicas (8)HABITUS EXTERIORAnomalías cutáneas (29)Anomalías oftalmológicas (10)Malformaciones y dismorfias (10)Anomalías esqueléticas (4)Anomalías cefálicas (3)SISTEMA INMUNOLÓGICOInfecciones recurrentes (27)Fiebre (17)Sepsis (17)Hipotermia (2)Síndrome de Reye (1)APARATO URINARIOOrina de olor extraño (25)Acidosis tubular renal (11)Insuficiencia renal (7) |
El número de pacientes que presentaron el signo o síntoma se enumera entre paréntesis.
La edad promedio de inicio de síntomas fue de 7,5 meses (mínimo 12 h de vida extrauterina y máximo 6 años) y la edad promedio al momento del diagnóstico fue de 2,1 meses (mínimo a los 7 días de vida y máximo 17,5 años); La correlación promedio entre la edad de inicio de los síntomas referidos por los padres y la edad del paciente al momento del diagnóstico bioquímico definitivo fue de 0,6 y en la figura 1 se muestra categorizada por grupo de edad. En promedio, el intervalo entre el inicio de los síntomas y el diagnóstico fue de 18 meses, con un mínimo de 3 días y un máximo de 16 años. El mínimo fue un caso de deficiencia de ornitina transcarbamilasa, cuyos síntomas fueron irritabilidad, somnolencia y deterioro neurológico súbito que inició a los 12 días de vida, requiriendo ingreso al servicio de urgencias de un hospital pediátrico en el cual se estableció la sospecha de un EIMi, enviando de manera inmediata las muestras de sangre y orina a nuestro centro diagnóstico.
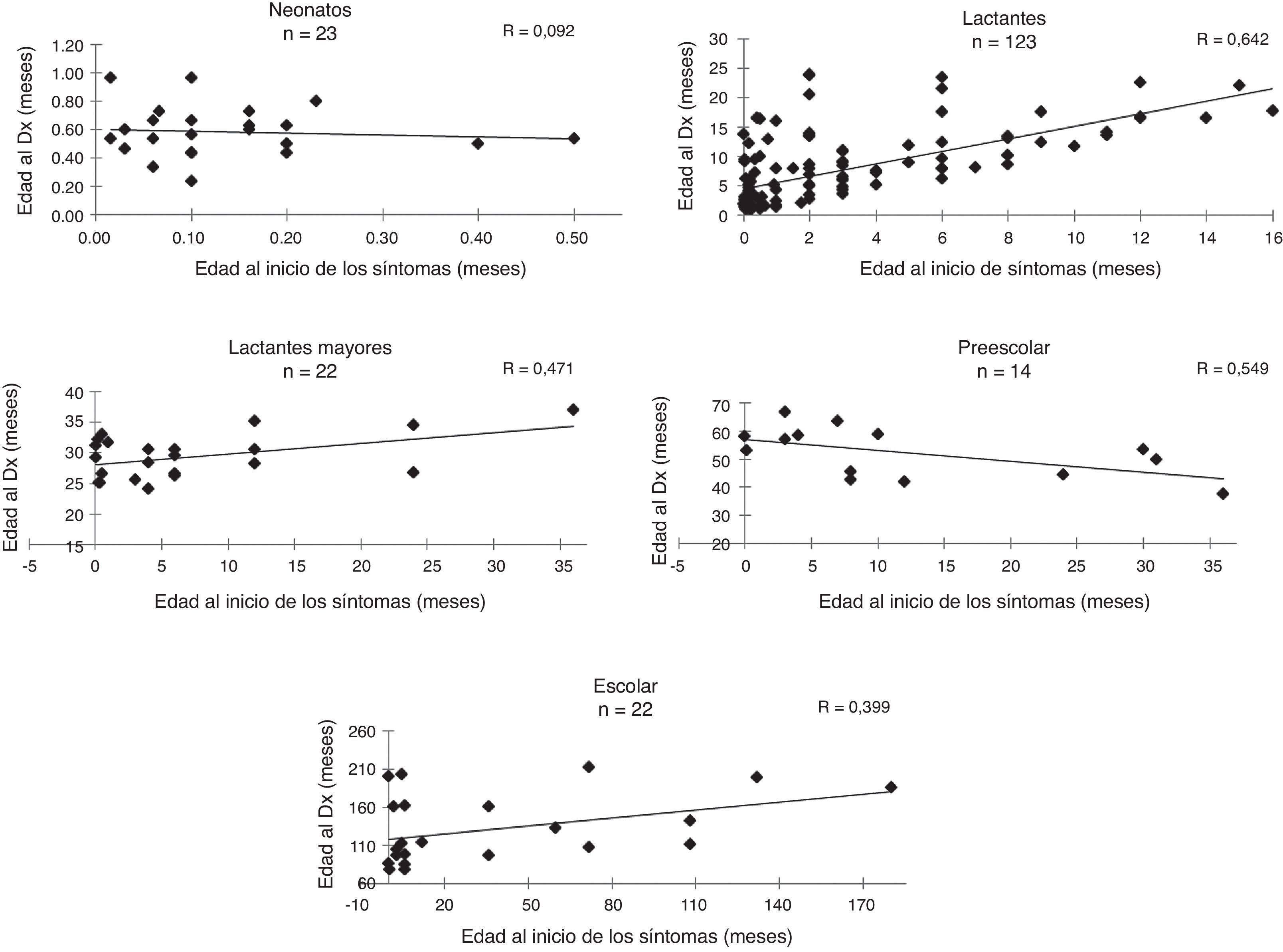
El intervalo máximo fue de 16 años: un niño con argininemia que presentó deterioro neurológico inexplicable en el periodo neonatal, ameritando hospitalización a los 11 días de vida y desarrollando posteriormente crisis convulsivas, diagnosticadas como síndrome de Lennox-Gastaut, con rechazo crónico selectivo a los alimentos altos en proteína, con detención global del desarrollo y retraso mental profundo, que fue valorado por varios especialistas; el motivo de referencia de este paciente fue la sospecha realizada por un pediatra general quien, después de recibir una capacitación sobre EIM, envió las muestras correspondientes.
DiscusiónLos EIMi son trastornos complejos y heterogéneos que representan un reto diagnóstico para los médicos, especialmente en aquellos países que no cuentan con programas de detección masiva neonatal y en los que hay pocos centros de referencia; además, estudios previos han documentado que las aminoacidopatías, las acidurias orgánicas y los defectos de la beta oxidación son los EIMi tratables que más comúnmente amenazan la vida de los pacientes pediátricos10,15,16.
Al igual que otros grupos que han estudiado EIMi mediante el tamiz selectivo10,15-18, encontramos una gran variedad de estas afecciones entre los niños enfermos mexicanos (25 tipos diferentes señalados en la tabla 1). En esta población analizada, las aminoacidopatías y las acidemias orgánicas fueron los EIMi más frecuentes. De las acidemias orgánicas, lo más observado fueron los trastornos del propionato (MMA y PA), que representaron el 68% de dicho grupo (69/102), lo cual coincide con lo descrito por otros autores15,18-22. De las aminoacidopatías, la más común fue la enfermedad de orina de jarabe de arce (30% de las aminoacidopatías y 15% de todos los EIMi). A diferencia de lo observado en poblaciones europeas, en las que las hiperfenilalaninemias son las aminoacidopatías más frecuentes y tienen una prevalencia de 1:12.363 recién nacidos23, estos defectos parecen ser más raros entre algunos grupos latinoamericanos, particularmente en México y en Cuba, donde se ha reportado una prevalencia al nacimiento de 1:96.400 y 1:52.590 recién nacidos, respectivamente24.
Los defectos del ciclo de la urea (citrulinemia, deficiencia de ornitina transcarbamilasa, acidemia argininosuccínica y argininemia) se encontraron en el 12,7% de todos los EIMi, por lo que también representan un grupo importante que se debe considerar.
En el presente trabajo, solo se encontraron 2 casos con defectos de oxidación de ácidos grasos, esto pudiera tener varias explicaciones: a) los pacientes fallecen súbitamente y el diagnóstico no se sospecha, por lo que no se realiza estudio metabólico alguno (autopsia bioquímica)25; b) presencia de falsos negativos para el análisis bioquímico, ya sea por depleción de carnitina o debido a que la muestra no se tomó al momento justo de la descompensación aguda26,27, y c) la prevalencia de estos trastornos es baja en nuestra población, al igual que lo documentado por otros grupos10,13,18.
Con la introducción de la MS/MS, se incrementó el número de casos diagnosticados en un 50% (tabla 2). Nuestros resultados muestran que el uso de una plataforma analítica consistente, compuesta por HPLC+GC/MS+MS/MS, mejora la detección diagnóstica de los EIMi dentro de la población de niños enfermos. Es importante mencionar que esto puede deberse tanto a la alta sensibilidad analítica del equipo, como a la capacidad para procesar un mayor volumen de muestras.
Las características clínicas y de laboratorio que hacen sospechar un EIM en niños enfermos son bien conocidas11,17,28 y coinciden con las halladas en estos pacientes (tabla 3). En nuestro estudio, encontramos que el cuadro clínico típico se caracteriza por irritabilidad, rechazo al alimento, vómito, hipotonía, trastornos del estado de alerta, retraso del neurodesarrollo o crisis convulsivas, acompañadas de alteraciones bioquímicas tales como acidosis metabólica de brecha aniónica aumentada e hiperamonemia.
A diferencia de los estudios previos realizados en México, en los cuales los pacientes eran enviados principalmente por genetistas y neurólogos29, registramos una mayor participación de los pediatras en la referencia de pacientes con EIMi. Esto probablemente responde a una mayor difusión del conocimiento sobre estas afecciones dentro del personal de salud mediante la impartición de conferencias, cursos y talleres; sin embargo, todavía debe reforzarse la capacitación puesto que la mayoría de los pacientes llegan a nuestro centro en forma tardía y en todos los grupos de edad estudiados el intervalo entre el inicio de los síntomas y el diagnóstico es inaceptablemente largo (fig. 1). Es sustancial que reconozcan tempranamente los datos clínicos y sepan dónde realizar pruebas de laboratorio especializadas o enviar a los pacientes a los centros de referencia.
En este grupo, cerca del 25% de las familias tuvieron antecedentes heredofamiliares positivos para un probable EIM, por lo que realizar el interrogatorio dirigido en relación con muertes inexplicables en la infancia y otros hijos afectados con un cuadro clínico similar al del propósito cobra gran relevancia en el abordaje diagnóstico.
Anecdóticamente, el padre de una paciente PKU refirió a 2 casos de la misma enfermedad, en los que reconoció las características de pelo rubio, retraso del desarrollo y crisis convulsivas basado en su propia experiencia y conocimiento.
La búsqueda intencionada de EIM mediante el diagnóstico o tamiz selectivo es especialmente importante en niños en los cuales no se llevó a cabo un TNA30, considerando que su limitante es que el paciente ya presentó una o varias descompensaciones metabólicas que pudieron dejar secuelas graves, especialmente a nivel neurológico. Existen evidencias sólidas, de buena calidad y orientadas a los pacientes, que establecen que el reconocimiento precoz de los EIM tiene el potencial de reducir la morbilidad y la mortalidad en los infantes afectados31,32 y que la MS/MS permite la identificación temprana de estas enfermedades en población asintomática31,33, por lo que lo ideal es hacer el TNA; sin embargo, en los países que no tienen establecidos dichos programas, ya sea por dificultades logísticas del sistema sanitario o por cuestiones económicas, el tamiz selectivo es una opción viable, recordando que el diagnóstico de estas enfermedades, incluso en el peor escenario —cuando el niño fallece—, es indispensable para otorgar el asesoramiento genético de certeza y plantear acciones preventivas en futuros embarazos, ya que la mayoría de estas afecciones hereditarias tienen riesgo de recurrencia del 25%11.
Estos resultados pueden servir como evidencia para que los EIMi sean incorporados al TNA en México o, en su defecto, se realice de manera sistemática el diagnóstico selectivo en todos los niños hospitalizados con datos clínicos sugestivos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a los médicos de todas las instituciones que tuvieron la confianza para enviar a sus pacientes al Instituto Nacional de Pediatría. También agradecemos la entusiasta participación de la Dra. Yamel Carolina Guevara Márquez en la captura de los datos clínicos y a la Q.F.B. Aída Hernández Montiel, por su colaboración en el procesamiento analítico de las muestras.
