




Introducción
La ausencia típica es un tipo de crisis epiléptica que consiste básicamente en una alteración de la consciencia, de breve duración, con descargas generalizadas de complejos punta-onda a 3 Hz en el electroencefalograma (EEG). Se dividen en simples y complejas, según se trate de una afectación exclusiva de la consciencia o bien se acompañen de síntomas motores, automatismos y/o signos vegetativos, respectivamente 1-4.
El nuevo esquema diagnóstico de las epilepsias y síndromes epilépticos de la International League Against Epilepsy (ILAE), dentro del grupo de epilepsias generalizadas idiopáticas, reconoce la epilepsia ausencia infantil como un síndrome epiléptico bien diferenciado, mientras que en el adolescente las crisis de ausencia junto con las mioclónicas y las tónico-clónicas quedan definidas como variables semiológicas de un continuum neurobiológico5. Se han descrito otras formas idiopáticas de epilepsia ausencias, tales como las ausencias con mioclonías periorales, las mioclonías palpebrales con ausencias y la epilepsia con ausencias mioclónicas, aunque tan sólo esta última ha sido considerada por la ILAE 3,5. Es decir, el diagnóstico sindrómico de las ausencias plantea dificultades con relativa frecuencia, ya que constituye una semiología ictal común a distintos síndrome epilépticos.
El objetivo del presente trabajo consiste en exponer y analizar las características epidemiológicas, clínicas y evolutivas de un grupo de pacientes con epilepsia ausencias en la edad pediátrica para facilitar su diagnóstico en la práctica clínica diaria.
Material y métodos
Se han revisado de forma retrospectiva 55 historias clínicas, de pacientes diagnosticados de ausencias típicas en la Unidad de Neuropediatría del Hospital Virgen del Camino de Pamplona entre enero de 1995 y diciembre de 2000. Para el diagnóstico y clasificación de las epilepsias ausencias se han aplicado los criterios actualizados de la ILAE 5 y Panayiotopoulos 3. Fueron rechazados 4 pacientes porque el seguimiento evolutivo puso de manifiesto que se trataba de un caso de epilepsia con crisis astatomioclónicas, otro de epilepsia benigna occipital, otro de crisis parciales complejas y, por último, uno de epilepsia mioclónica juvenil.
De cada historia clínica se registraron datos epidemiológicos: sexo, edad al diagnóstico, antecedentes personales (embarazo y parto, período neonatal, desarrollo psicomotor y convulsiones febriles), antecedentes familiares de epilepsia; datos clínicos: motivo de consulta, frecuencia y tipo de crisis (simples o complejas), junto con exámenes complementarios: EEG en reposo y tras activaciones (hiperventilación y estimulación luminosa intermitente) y estudios de neuroimagen (tomografía computarizada [TC] y/o resonancia magnética [RM] craneal), y datos evolutivos: fármacos antiepilépticos empleados, respuesta clínica y del EEG al tratamiento, supresión farmacológica, recaídas y tiempo de seguimiento.
Tras el diagnóstico, la totalidad de pacientes incluidos en la muestra fueron citados cada 15 días hasta la remisión de las crisis y, posteriormente, salvo cambios evolutivos significativos, a los 3, 6 y 12 meses, y luego cada año. En cada consulta se realizó EEG.
Los resultados se expresan como medias con sus desviaciones estándar y como porcentajes. Para el análisis estadístico (t de Student, comparación de proporciones) se utilizó el programa informático Sigma-Plus (Hardware, 97).
Resultados
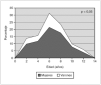
El tiempo medio de seguimiento evolutivo de estos pacientes ha sido de 6,9 ± 2 años (intervalo: 4,8-11). La edad media en el momento del diagnóstico era de 7,5 ± 2,7 años (intervalo: 2,5-13,2), mientras que la edad media actual es de 13,9 ± 3,2 años (intervalo: 7,6-19,3). Existía una mayor prevalencia del sexo femenino (72,5 %) respecto al masculino (27,5 %) sin que existieran diferencias significativas en la edad media de diagnóstico entre ambos sexos (varones: 7,6 ± 2,8 años; mujeres: 7,2 ± 2,6 años). Las edades correspondientes al diagnóstico seguían una curva de distribución normal con un pico máximo en el intervalo de edad de 6 a 8 años (fig. 1). El 70,6 % de los casos fueron diagnosticados en edad escolar; mientras que en edad preescolar y en la adolescencia lo fueron el 13,7 y el 15,7 % de los casos, respectivamente. Por tanto, según los criterios de la ILAE, se podría hablar de epilepsia ausencia infantil en 43 casos (84,3 %) y de epilepsia ausencia juvenil en 8 casos (15,7 %).
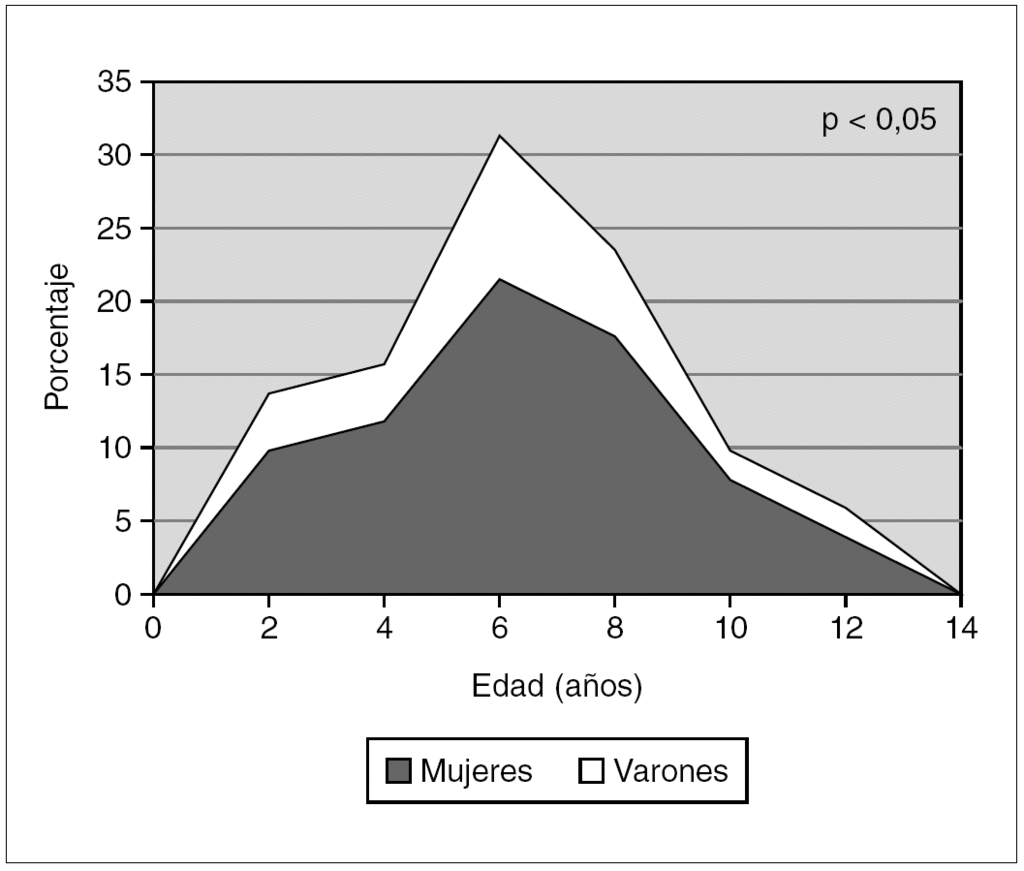
Figura 1. Distribución de la edad en el momento del diagnóstico.
La totalidad de pacientes procedían de embarazos a término. El 81,9 % de los partos fueron eutócicos y el 18,1 % terminaron instrumentados y/o por cesárea. Los pesos al nacer oscilaron entre 2.450 y 4.250 g. El período neonatal y desarrollo madurativo fueron normales en la totalidad de casos, salvo en una niña con síndrome de Down. En 8 casos existían antecedentes personales de convulsiones febriles (15,7 %). La historia familiar de epilepsia era positiva en 16 casos (32 %), bien en parientes de primer grado (n = 4) o de segundo grado (n = 12). En 8 casos (16 %) había antecedentes familiares de ausencias, y entre ellos 2 casos eran hermanas.
El motivo de la consulta en el 94,1 % de los casos fue la sospecha de crisis repetidas de alteración del nivel de consciencia o desconexiones con una frecuencia variable, desde algunas crisis semanales y/o mensuales (20 %) hasta decenas en un mismo día (80 %) y que, en algunos casos (9,8 %), eran más frecuentes en situaciones de cansancio y/o estrés. Un caso (varón de 11,3 años) acudió a urgencias por presentar un estado de confusión y/o desorientación y cuyo EEG puso de manifiesto ausencias subintrantes (estatus) que cedieron con benzodiazepinas. En 8 casos (se excluye la niña con síndrome de Down) se asociaba una disminución del rendimiento escolar, y otros 2 casos (3,9 %) consultaron exclusivamente por este motivo y fue al realizarse un EEG cuando se objetivaron las ausencias. El tiempo medio de evolución en el momento del diagnóstico fue de 3,5 meses (intervalo: 0,5-24); pero en 5 casos oscilaba entre 6 y 8 meses, y en otros 5 casos entre 12 y 24 meses.
En el 51 % de los casos (n = 26) las ausencias eran simples y consistían exclusivamente en una interrupción brusca de la actividad y mirada fija (18 casos) que podía acompañarse de parpadeo rítmico (8 casos). Mientras que en el 49 % de los casos (n = 25) las ausencias eran complejas y se acompañaban de ligeras contracciones de hombros (2 casos), caída de cabeza (un caso), retropulsión del tronco (un caso), lateralización cefálica y desviación ocular (2 casos), desviación de la mirada (6 casos), movimientos de chupeteo o deglución (10 casos), marcha automática (un caso) o relajación de esfínteres (2 casos). Dos pacientes, diagnosticados a los 10,5 y 11,8 años de edad, presentaron crisis tónico-clónicas generalizadas, uno antes y otro después del diagnóstico y tratamiento. De los 43 casos de epilepsia ausencia infantil, 23 tenían ausencias simples (53,5 %) y 20 complejas (46,5 %); mientras que de los 8 casos de epilepsia ausencia juvenil, tres tenían ausencias simples (37,5 %) y cinco complejas (62,5 %).
En el EEG ictal (fig. 2) la actividad de fondo era normal en todos los casos, y el trazado se caracterizaba por ráfagas generalizadas de complejos punta-onda (84,3 %) y polipuntas-onda (15,7 %) con una frecuencia de 3 Hz en el 88,1 % de los casos (intervalo: 2,5-4) que, ocasionalmente, coexistían con puntas-ondas en regiones frontales (un caso), centrales (2 casos) o posteriores (3 casos). Salvo en un caso, la hiperventilación intensificaba estos hallazgos (98 %). Existía un correlato electro-clínico en el 81,4 % de los casos (en reposo y/o tras hiperventilación). La estimulación luminosa intermitente era positiva en 8 casos (15,7 %), aunque en todos ellos la hiperventilación también era positiva. La duración media de los episodios de ausencias era de 12 s (intervalo: 4-35). El 18,6 % no llegaban a 5 s, el 69,7 % oscilaban entre 5 y 20 s y el 11,6 % superaban los 20 s; no obstante, el 53,7 % no alcanzaban los 10 s. Se realizaron estudios de neuroimagen en el 90,2 % de los casos: TC craneal (54,3 %) o RM craneal (45,7 %) con resultados normales, salvo en un caso que presentaba un quiste de la fosa posterior comunicado con el IV ventrículo (variante de Dandy-Walker).
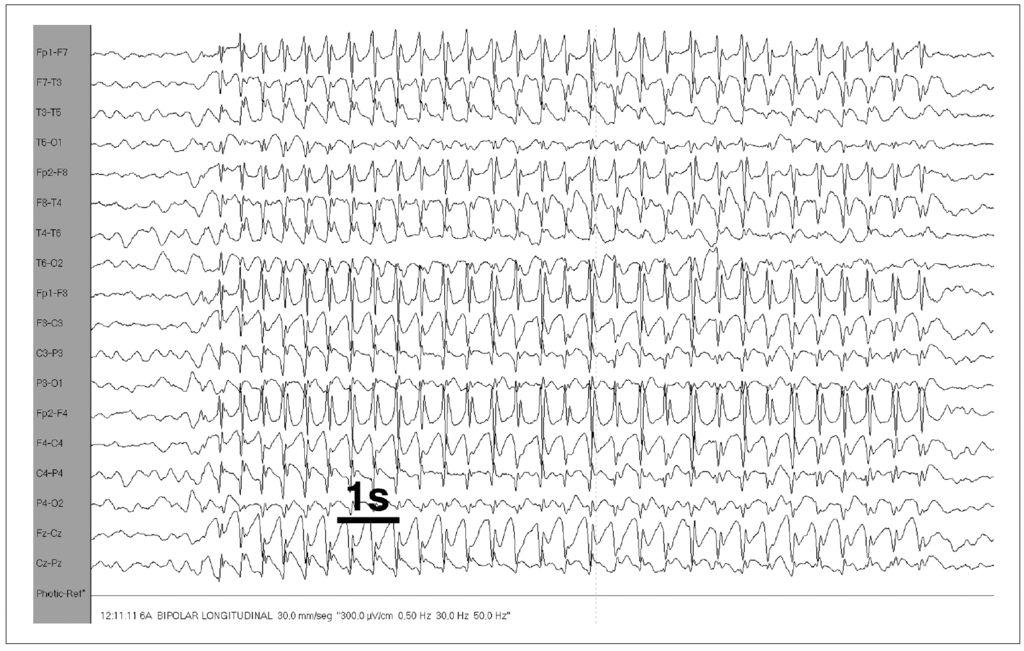
Figura 2. EEG ictal en una niña de 8,5 años: complejos punta-onda generalizados a 3 Hz y con una duración aproximada de 12 s.
El fármaco utilizado inicialmente fue el ácido valproico (VPA), a una dosis media de 26 mg/kg/día (intervalo: 18,6-36,4). En la figura 3 se exponen los valores porcentuales de la respuesta clínica y del EEG al tratamiento. Una vez iniciado, a los 3 meses el 84,3 % de los casos estaban libres de crisis y el 70 % de los EEG se habían normalizado, existiendo una correlación lineal positiva (r = 0,71) y estadísticamente significativa (p < 0,05) entre ambos parámetros. El tiempo medio de la remisión de las crisis fue de 1,6 meses, y el tiempo medio de normalización del EEG, de 2,6 meses. En 8 casos (15,7 %) las crisis de ausencias seguían a los 3,5 meses de iniciado el tratamiento. El VPA fue sustituido por etosuximida en 6 casos, a una dosis media de 22,7 mg/kg/día (intervalo: 21-25), y en los otros 2 casos se asoció clonazepam, en uno de ellos tras presentar un exantema cutáneo secundario a la etosuximida. En estos casos, el tiempo medio de remisión de las crisis tras la prescripción de etosuximida y/o clonazepam fue de 1,3 meses, y el tiempo medio de normalización del EEG de 1,8 meses. El tratamiento fue retirado en 43 casos (84,3 %), tras una duración media de 3,3 ± 0,6 años (intervalo: 2,2-5,1). Tras su retirada y tras un seguimiento medio de 3,5 años (intervalo: 2,1-7,3) 3 casos (uno diagnosticado en edad escolar y dos en la adolescencia) recayeron a los 2, 4 y 5 meses, respectivamente; y en todos ellos se prescribió VPA con buena respuesta terapéutica hasta la actualidad. En 8 casos (15,7 %) no se ha llegado a retirar la medicación (2 diagnosticados en edad preescolar, 3 escolares y 3 adolescentes) por presentar a lo largo del control evolutivo crisis aisladas y/o EEG con complejos punta-onda tras hiperventilación. La duración media del tratamiento en estos pacientes fue de 7,3 años (intervalo: 4,5-10,5). De estos 8 casos, cuatro habían tenido una pobre respuesta al tratamiento inicial con VPA y hubo que sustituirlo por etosuximida (2 casos) o asociar clonazepam (2 casos), y los otros 4 casos habían sido tratados de manera ininterrumpida con VPA.
Figura 3. Respuesta clínica y del EEG al tratamiento antiepiléptico.
De los 10 casos (19,6 %) con afectación del rendimiento escolar (se excluye la niña con síndrome de Down), nueve mejoraron con la respuesta clínica y el otro, a pesar de una favorable evolución y retirada de la medicación, mantuvo un retraso de aprendizaje y alteración conductual. Además, 3 casos (5,9 %) presentaron durante el control evolutivo un deterioro del rendimiento escolar que mejoró tras suprimir la medicación: VPA, etosuximida y VPA-clonazepam, respectivamente.
Discusión
Las epilepsias generalizadas idiopáticas suponen cerca de un tercio de las epilepsias infantiles 3,6 y conforman un grupo heterogéneo de síndromes epilépticos posiblemente con patrones genéticos y mecanismos patogénicos diferentes que, aunque pueden darse a cualquier edad, tienen una mayor expresividad en la edad escolar y adolescencia. Las características diferenciales entre los distintos síndromes epilépticos son difíciles de establecer debido al solapamiento semiológico que puede darse entre ellos y, de hecho, el control evolutivo de estos pacientes suele inducir diferentes diagnósticos sucesivos que sugieren un continuum neurobiológico. Entre las epilepsias generalizadas idiomáticas, la epilepsia ausencia es el tipo más frecuente, y llega a representar el 6-10 % de las epilepsias en la edad pediátrica 6-9.
La delimitación de las epilepsias ausencias entre forma infantil y juvenil es muy controvertida, e incluso algunos autores hacen referencia a las ausencias que aparecen en la edad preescolar como un subgrupo diferenciado con un pronóstico variable 10. Conviene tener en cuenta que las ausencias, aparte de conformar un síndrome epiléptico específico en la infancia, pueden suponer la expresión inicial de distintos síndromes epilépticos. Por tanto, el control evolutivo es esencial para corroborar o no el diagnóstico inicial, tal como ocurrió en esta serie y que obligó a la exclusión de 4 casos. Al no existir marcadores biológicos específicos, se ha llegado a proponer una clasificación basada estrictamente en criterios clínicos que distinguiría, por ejemplo, entre ausencias puras, ausencias con crisis tónico-clónicas, ausencias fotosensibles, etc. 11. En este trabajo se ha seguido la nomenclatura de la ILAE que distingue entre ausencias infantiles y juveniles; aunque en su versión recientemente actualizada se define a la epilepsia ausencia juvenil, junto a la epilepsia mioclónica y a las crisis tónico-clónicas generalizadas, como variables fenotípicas de las epilepsias generalizadas idiopáticas en la adolescencia 5. Como el tamaño de esta serie no permitía un estudio comparativo entre ambos subgrupos, para facilitar el análisis estadístico se han englobado todos los casos en un grupo común, aunque haciéndose algunos comentarios sobre algunas diferencias observadas entre ellos.
Entre los aspectos epidemiológicos de esta serie cabe destacar que la edad correspondiente al momento del diagnóstico seguía una distribución normal, con un pico de máxima incidencia en la edad escolar y una sensible preponderancia del sexo femenino frente al masculino (cociente: 2,6), lo que coincide con los datos publicados 1,3,6-8. Por tanto, la epilepsia ausencia parece tratarse de una patología específicamente pediátrica y que afectaría preferentemente a niñas en edad escolar. Cabe apuntar que el 13,7 % de los casos fueron diagnosticados en edades tempranas, entre los 2,5 y 3,9 años de edad, sin que por ello hayan tenido un peor pronóstico; quizá la pretensión de englobar estos casos como un subgrupo de pronóstico variable se deba a que a menor edad la expresividad clínica sería más versátil y, por tanto, el diagnóstico sindrómico más controvertido.
La elevada prevalencia de antecedentes familiares con trastornos paroxísticos registrados en estos pacientes sugiere una base genética, probablemente de tipo poligénico y/o multifactorial 2,12,13. En esta serie el 32 % de los pacientes contaban con antecedentes epilépticos, y en la mitad de los casos se trataba de familiares de segundo grado, salvo el caso de las 2 hermanas ya citadas, que habían padecido ausencias en su infancia.
La duración de las ausencias suele oscilar entre 5 y 15 s y, de hecho, en esta serie la duración media fue de 12 s. No obstante, en la mayoría de casos las crisis eran más breves, sin alcanzar siquiera los 10 s, e incluso infraclínicas (< 5 s), lo que explicaría que pasaran inadvertidas y, en consecuencia, la sospecha diagnóstica se retrasara sensiblemente, como ocurrió en esta serie en un porcentaje bastante importante de casos.
La posibilidad de registrar de forma simultánea las características clínicas y electroencefalográficas con vídeo-EEG facilita tanto su clasificación semiológica como el diagnóstico sindrómico 1-4. De hecho, en esta serie la aplicación de esta técnica permitió, en gran medida, distinguir entre ausencias simples y complejas; porque en la mayoría de los casos los padres referían haber observado en sus hijos ausencias aparentemente simples. Es decir, episodios de supresión brusca de la actividad y desconexión sensorial, en los que el niño parecía estar distraído, y con la mirada perdida o ligero aleteo palpebral, de duración breve y con reanudación inmediata de la actividad que estaba realizando como si nada hubiera pasado, aunque sin tener consciencia de lo ocurrido durante las crisis. Pero fue precisamente la monitorización vídeo-EEG lo que permitió objetivar y analizar la asociación de síntomas motores clónicos, tónicos o atónicos, automatismos o signos autonómicos y, en consecuencia, establecer de manera definitiva el tipo de ausencias.
El patrón EEG característico de estos pacientes muestra una actividad basal bien estructurada con descargas generalizadas ictales de complejos punta-onda y/o polipuntas-onda (< 3 puntas) a 3 Hz, bilaterales, simétricas y síncronas 1-4. La hiperventilación suele inducir la aparición de ausencias electroclínicas, de tal manera que algunos autores consideran su negatividad como un criterio de exclusión, o al menos de reconsideración diagnóstica 11. En esta serie existía un correlato electroclínico en el 81,4 % de los casos, bien en condiciones basales y, en su defecto, tras hiperventilación. La estimulación luminosa intermitente era positiva en el 15,7 % de los casos, aunque en todos ellos la hiperventilación también era positiva.
Aunque clásicamente se consideraba que las ausencias afectaban de manera exclusiva a niños sin problemas cognitivos, se admite que el 2-6 % de los casos presentan patología neurológica previa, lo que suele ir asociado con refractariedad terapéutica 1,11,14. En esta serie se incluyen 2 casos (3,9 %) con problemas cognitivos: el caso de una niña con síndrome de Down, diagnosticada a los 1,3 años de ausencias simples, con RM craneal normal y buena respuesta al VPA y que fue retirado a los 36 meses de evolución; en la actualidad, con 7,6 de edad, está asintomática. Y el caso de un varón con retraso de aprendizaje, diagnosticado a los 6,8 años de ausencias simples, con una variante de Dandy-Walker, y con buena respuesta al VPA que fue retirado a los 32 meses de evolución, y que en la actualidad, con 15,6 años, está asintomático, pero sigue con retraso escolar y problemas conductuales. En un porcentaje relativamente importante de casos, quizás en relación con el número de ausencias diarias, referían disminución del rendimiento escolar que se normalizaba de manera simultánea con la respuesta terapéutica; no obstante, conviene tener presente la posibilidad de deterioro cognitivo como efecto colateral de los fármacos antiepilépticos 15,16, como se constató en 3 casos.
No existen datos lo bastante contrastados para decantarse entre VPA, etosuximida y lamotrigina en el tratamiento de las ausencias 17; sin embargo, el uso del VPA como fármaco de primera elección está ampliamente reconocido 1-4,8,16. En esta serie, con el VPA en monoterapia se controlaron el 84,3 % de los pacientes; con tiempos medios de remisión de las crisis y normalización del EEG de 1,6 y 2,6 meses, respectivamente. Con tratamiento, la evolución y pronóstico, tanto en las formas infantiles como juveniles, son muy favorables, con una adaptación familiar y/o social del todo normal. El planteamiento de la retirada de los fármacos antiepilépticos es controvertido y depende del tipo de síndrome epiléptico que se padezca; aunque, en general, se exige un período libre de crisis superior a 2 años y la normalización del trazado electroencefalográfico. En el caso de las ausencias, salvo criterios desfavorables 18,19, tras retirar la medicación las recidivas, al menos a corto y/o medio plazo, son relativamente escasas, aunque habría que ser muy prudentes a largo plazo, en especial en las formas juveniles 3,20. En esta serie se retiró la medicación en un tiempo medio de 3,3 años a 43 pacientes y tras un seguimiento medio posterior de 3,5 años han recaído tres (7 %) a los pocos meses de la retirada del fármaco.
En suma, las ausencias constituyen un síndrome epiléptico relativamente frecuente en la edad pediátrica, sobre todo en la edad escolar, que pueden pasar inadvertidas y, en consecuencia, condicionar el rendimiento académico. La respuesta al tratamiento es excelente y su pronóstico, bueno. No obstante, como podrían ser la expresión inicial de distintos síndromes epilépticos sería obligado mantener un riguroso control evolutivo de estos pacientes.
Correspondencia: Dr. T. Durá Travé.
Avda. Pío XII, 10, 8.ºC. 31008 Pamplona. España.
Correo electrónico: tduratra@cfnavarra.es
Recibido en mayo de 2005.
Aceptado para su publicación en julio de 2005.
