





Conocer la incidencia de la atrofia espinal infantil (AME) en nuestra población y estudiar la distribución genética y las características epidemiológicas y clínicas, el nivel de cuidados y su evolución.
Material y métodoEstudio descriptivo retrospectivo de los pacientes atendidos en nuestro hospital en los últimos 25 años (1987-2013), con diagnóstico clínico y neurofisiológico de AME.
ResultadosSe halló a 37 pacientes, lo que supone una incidencia aproximada de 1/10.000 recién nacidos vivos. Predominaba el sexo masculino (relación varón/mujer: 1,6/1). El tipo de AME diagnosticado más frecuentemente fue el tipo i (26 casos), seguido del tipo ii (9 casos), un caso de AME tipo iii, y otro caso de spinal muscular atrophy with respiratory distress type 1 (SMARD 1). La alteración genética más frecuente fue la deleción en homocigosis de exones 7 y 8 del gen SMN1, en 31 casos, mientras que 5 pacientes presentaban una genética atípica. La mediana de supervivencia para el tipo i fue de 8,0 meses y de 15,8 años para el tipo ii.
ConclusionesLa incidencia en nuestra población permanece estable en torno a 1/10.000. La mayoría de los casos presenta una genética típica con predominio de varones. En aproximadamente 1/10 pacientes la alteración genética fue diferente de la clásica. La prevalencia de AME no relacionadas con el gen SMN fue de 1/37. El nivel de cuidados se ha incrementado en los últimos años, en consonancia con las demandas sociales y asistenciales.
To determine the incidence of spinal muscular atrophy (SMA) in our study population and genetic distribution and epidemiological and clinical characteristics and to analyze the level of care and development.
Material and methodRetrospective descriptive study of patients treated in our hospital in the past 25 years (from 1987 to early 2013), with a clinical and neurophysiological diagnosis of SMA.
ResultsA total of 37 patients were found, representing an incidence for our reference population and year of 1 case per 10,000 live births. Males predominated (male/female ratio: 1.6/1). The type of SMA diagnosed more frequently was, type i (26 cases), followed by type ii (9 cases), one case with SMA type iii, and one case of spinal muscular atrophy with respiratory distress type 1 (SMARD1). The most frequent genetic alteration was homozygous deletion of exons 7 and 8 of SMN1 gene in 31 cases, while five patients had atypical genetics. The median survival for type i was 8.0 months and 15.8 years for type ii.
ConclusionsThe incidence in our population remains stable at around 1/10.000. Most cases presented with, predominantly male, typical genetics. In approximately 1/10 patients the genetic alteration was different from the classical one to the SMN gene. The prevalence of AME unrelated SMN gene was 1/37. The level of care has increased in line with social and welfare demands in recent years.
Desde las primeras descripciones por Werdnig, en 1891, y Hoffmann, en 1893, mucho se ha avanzado en el conocimiento de la atrofia muscular espinal (AME). Conocemos que se debe a la deficiencia en la traducción de la proteína de la supervivencia de la motoneurona (SMN), que parece estar involucrada en varias funciones esenciales para la célula (metabolismo del ARN, su procesamiento y empalme) y otras que tienen que ver más específicamente con la supervivencia de las neuronas motoras alfa (apoptosis, transporte axonal) en el asta anterior de la médula espinal. La proteína SMN está codificada por los genes SMN1 y 2. La incidencia mundial descrita es de aproximadamente 1/10.000 nacimientos, siendo portadoras 1 de cada 40 personas.
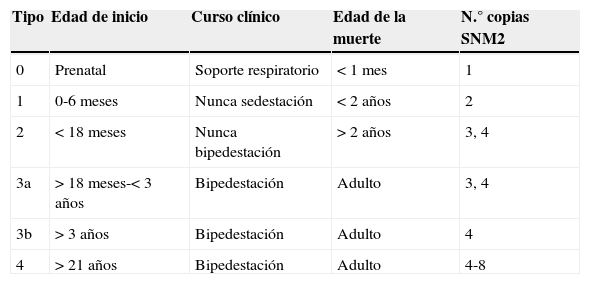
En la forma clásica, la semiología por la que se caracteriza es la debilidad y la atrofia muscular de predominio proximal, de comienzo y afectación en grado variable según el tipo clínico de la AME. Clásicamente, se han distinguido 3 tipos: el infantil, el juvenil y la forma adulta. Actualmente, el Consenso Internacional sobre AME1,2 la clasifica según la clínica y la edad de inicio en más tipos, dividiendo en subtipos el tipo 3, según la edad de aparición. Se añade un tipo 4 para los casos de inicio en el adulto y se incluye un tipo 0 para los de inicio prenatal, que fallecen en las primeras semanas de vida (tabla 1). Existe variabilidad clínica entre individuos dentro de cada tipo y hasta un 25% de los pacientes no pueden catalogarse, pero esta clasificación es útil a nivel clínico y pronóstico.
Clasificación de la atrofia muscular espinal
| Tipo | Edad de inicio | Curso clínico | Edad de la muerte | N.° copias SNM2 |
|---|---|---|---|---|
| 0 | Prenatal | Soporte respiratorio | < 1 mes | 1 |
| 1 | 0-6 meses | Nunca sedestación | < 2 años | 2 |
| 2 | < 18 meses | Nunca bipedestación | > 2 años | 3, 4 |
| 3a | > 18 meses-< 3 años | Bipedestación | Adulto | 3, 4 |
| 3b | > 3 años | Bipedestación | Adulto | 4 |
| 4 | > 21 años | Bipedestación | Adulto | 4-8 |
El gen de la AME descubierto en 19953 asienta en el brazo largo del cromosoma 5 (5q11.1-13.3). Este gen ha recibido el nombre de SMN por «survival motor neuron». Los humanos tienen 2 copias casi idénticas que han sido llamadas SMN1 y SMN2. Existe una sola diferencia en el nucleótido a principios del exón 7 (C para el SMN1 y T para el SMN2) que es importante para el empalme o splicing del SMN-ARN. Se desconoce por qué esta proteína ubicua provoca esta enfermedad neuronal tan selectiva. Las deleciones del gen SMN1 son responsables de la AME. Alrededor del 95% de los pacientes con AME son homocigotos para la ausencia de los exones 7 y 8 del gen SMN1, y alrededor del 5% son heterocigotos compuestos para ausencia de exones 7 y 8 de un alelo SMN1 y una mutación puntual en el otro alelo SMN1. Las deleciones del gen SMN2 no son causa de enfermedad, sino que el número de copias del SMN2, que según los casos pueden variar, modula el fenotipo condicionando la gravedad de la AME, de forma que a mayor número de copias de SMN2 menor gravedad, aunque existen otros factores implicados4,5. En este sentido, los supuestos teóricos abordan la posibilidad de tratamiento genético de la enfermedad a través de una sobreexpresión inducida del gen SMN2 y puede no estar lejos el día en que se pueda encontrar mejoría para esta terrible enfermedad6,7. También ha tenido éxito experimental la terapia génica de introducción, mediante vectores virales, del gen de la proteína de la SMN en el genoma del modelo murino de la enfermedad8.
Otras formas con una clínica superponible a la AME, con matices, aparecen asociadas a una genética diferente. Se han descrito casos ligados a alteraciones del cromosoma 11: SMARD1, del inglés spinal muscular atrophy with respiratory distress type 19,10 y otros de forma recesiva ligada al cromosoma X, que comienza precozmente en varones, conocida como enfermedad de Kennedy11,12.
El objetivo de este trabajo es actualizar la incidencia de la AME en nuestra población, conocer la distribución genética y las características epidemiológicas y clínicas en los últimos 25 años a la luz de los conocimientos actuales, así como analizar el nivel de cuidados y evolución de la AME en nuestro medio, en relación con el consenso internacional1,2.
Material y métodosRealizamos un estudio descriptivo retrospectivo, mediante la revisión de las historias clínicas de pacientes valorados en nuestro hospital en los últimos 25 años, desde el año 1987 hasta inicios del 2013, con diagnóstico clínico y neurofisiológico de atrofia muscular espinal.
Se recogieron variables epidemiológicas, clínicas, genéticas, tratamiento de soporte recibido y supervivencia. Las variables recogidas fueron la edad al inicio de los síntomas y la edad al fallecimiento, el género, los antecedentes familiares con afectados previos, los síntomas iniciales, los estudios neurofisiológicos, los estudios genéticos, el tipo de AME, así como los estudios de diagnóstico prenatal posteriores, el tratamiento de soporte recibido y la supervivencia.
El estudio molecular se realizó a partir del ADN por reacción en cadena de la polimerasa con amplificación de los exones 7 y 8 del gen SMN1 en sangre de pacientes con sospecha de AME. Los genes SMN1 y SMN2 se diferenciaron por análisis de polimorfismo de longitud de fragmentos de restricción. Se interpretó como deleción homocigota del exón 7 del gen SMN1 la falta de uno de los 3 fragmentos de restricción. La deleción homocigota del exón 8 del mismo gen se evidenció por la ausencia de la banda de 189 pares de base sin digerir del gen SMN1, junto con la presencia de los 2 fragmentos de restricción del gen SMN2.
En los casos sin deleción en homocigosis, se cuantificó el número de copias del gen SMN mediante la técnica multiplex ligation-dependent probe amplification: al detectar una sola copia de SMN1 (porque la otra copia puede estar inactivada por mutación), se realizó un rastreo de la secuencia codificante del gen para identificar posibles deleciones, duplicaciones y mutaciones puntual.
ResultadosResultados epidemiológicos generalesSe halló a 37 pacientes que cumplían los criterios de inclusión, lo que supone una incidencia aproximada para nuestra población de referencia y año de 1,41 casos por cada 15.000 recién nacidos vivos (un caso por cada 10.638 recién nacidos vivos). Entre 1987 y 1999, fueron diagnosticados 14 pacientes, mientras que entre los años 2000 y 2013 los pacientes diagnosticados ascendían a 23, lo que supuso un claro aumento de los casos diagnosticados. Los diagnósticos de casos previos al año 1995 se hicieron a través de la constatación de familiares portadores en 3 casos y en otros 3 se trató de pacientes cuya genética se estudió con posterioridad al tratarse de AME tipo ii en 2 pacientes y AME tipo iii en otro.
La mayoría de los casos procedían de nuestra área de referencia (31/37). Predominó el sexo masculino, con 23 pacientes varones y una relación varón/mujer 1,6/1.
La edad media de inicio de la sintomatología fue de 2,2 meses en la AME tipo i (rango entre los 0 y 7 meses) y de 11,2 meses en el tipo ii (rango entre los 6 y 18 meses). Por otra parte, el único caso diagnosticado de AME tipo iii tenía 12 meses al inicio de los síntomas de enfermedad y el paciente diagnosticado de AME con distrés respiratorio (SMARD1) inició la sintomatología el primer mes de vida.
Resultados clínicosEl tipo de AME más frecuente fue el tipo i, en 26 casos, seguido del tipo ii en 9 casos. Un caso fue diagnosticado de AME tipo iii y otro caso SMARD1. No hubo casos con posible herencia ligada al X (fig. 1).

El síntoma inicial en la mayoría de los pacientes fue la hipotonía, en 27/37 casos, seguido del retraso motor en 5/37 casos. En 4 casos de AME tipo i se asoció trastorno en la succión-deglución, de los cuales 2 presentaron episodios de atragantamiento y otro caso de AME tipo i fue diagnosticado a raíz de un episodio de riesgo vital aparente con pausas de apnea. Ninguno de nuestros pacientes comenzó con artrogriposis ni con criterios de AME tipo 0. En 3 pacientes, el síntoma inicial fue distrés respiratorio asociado a infección intercurrente y en 2 fue trastornos de la marcha (fig. 2).

Se contabilizaron 92 embarazos en el total de las familias, entre los cuales constaban 9 casos de abortos espontáneos y un caso de interrupción voluntaria del embarazo. Los 9 casos ocurrieron en las familias de 18 pacientes afectados de AME tipo i seguidos en nuestro centro (en las que se contabilizaron 47 embarazos, lo que supone una tasa de aborto espontáneo de 19,1% en dicho grupo).
En 7/37 existían antecedentes familiares de AME y debilidad muscular no filiada en 3/37. En 4/37 casos existía antecedente de muerte neonatal: 3 de ellos eran hermanos de uno de los pacientes, el cual comenzó a los 8 días de vida con hipotonía, y fallecieron en el período neonatal precoz. Los 23/37 casos restantes no presentaban antecedentes familiares de enfermedades neurológicas o sintomatología relacionada (fig. 3).
Estudios genéticos
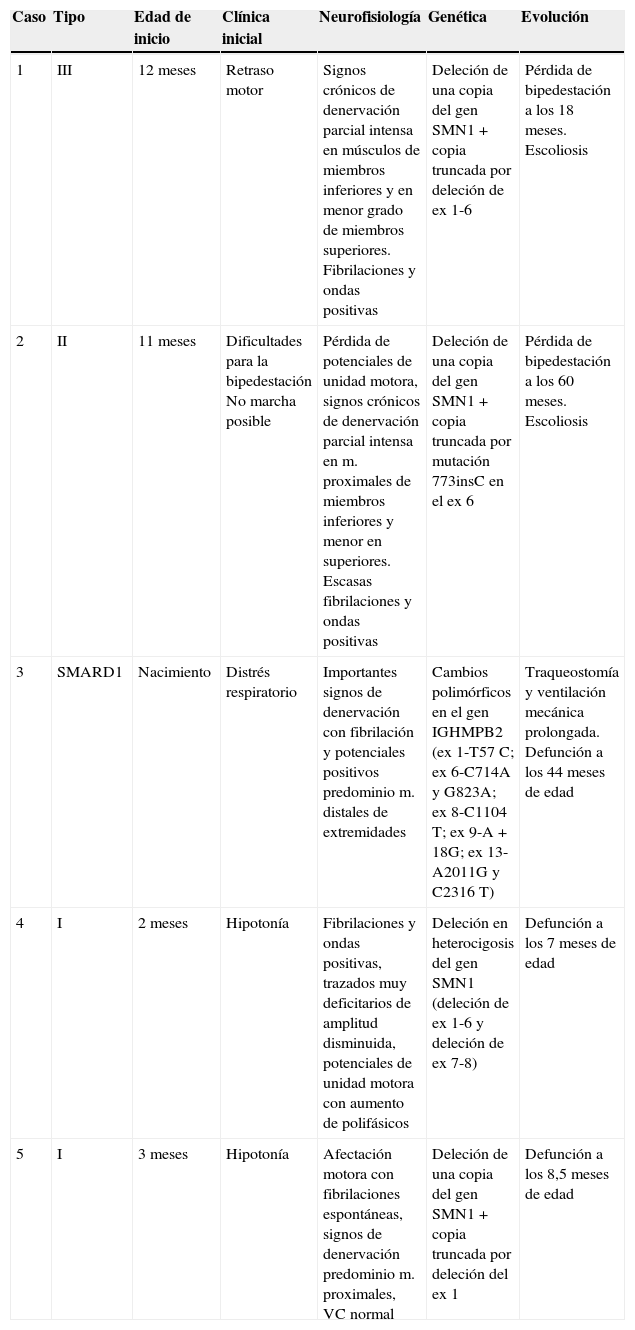
El estudio genético se realizó en 36 pacientes. En la mayoría de los casos, no fue posible evaluar el número de copias del gen SMN2. La alteración genética más frecuente fue la deleción en homocigosis de exones 7 y 8 del gen SMN1, en 31/36 casos, mientras que 5/36 pacientes presentaban una genética atípica. En 4 casos se detectó heterocigosis compuesta con deleción clásica de una copia del gen SMN1 y una copia truncada no funcional por diferentes mutaciones: en 2 casos por deleción de exones 1-6, en otro por deleción del exón 1 y un tercer caso por mutación 773insC. Por último, el paciente diagnosticado de AME con distrés respiratorio presentaba cambios polimórficos en el gen IGHMPB2 del cromosoma 11 (ex 1-T57C; ex 6-C714A y G823A; ex 8-C1104T; ex 9-A+18G; ex 13-A2011G y C2316T).
Las características epidemiológicas, clínicas, neurofisiológicas y evolutivas de los 5 casos de genética atípica se resumen en la tabla 2. En 6 familias se realizó diagnóstico genético prenatal en embarazos posteriores (16,2%) y se detectaron casos afectados en 4 gestaciones, casos portadores en 2 y casos sanos no portadores en otras 2 gestaciones (fig. 4).
Características epidemiológicas, clínicas, neurofisiológicas y evolutivas de los casos de genética atípica
| Caso | Tipo | Edad de inicio | Clínica inicial | Neurofisiología | Genética | Evolución |
|---|---|---|---|---|---|---|
| 1 | III | 12 meses | Retraso motor | Signos crónicos de denervación parcial intensa en músculos de miembros inferiores y en menor grado de miembros superiores. Fibrilaciones y ondas positivas | Deleción de una copia del gen SMN1+copia truncada por deleción de ex 1-6 | Pérdida de bipedestación a los 18 meses. Escoliosis |
| 2 | II | 11 meses | Dificultades para la bipedestación No marcha posible | Pérdida de potenciales de unidad motora, signos crónicos de denervación parcial intensa en m. proximales de miembros inferiores y menor en superiores. Escasas fibrilaciones y ondas positivas | Deleción de una copia del gen SMN1+copia truncada por mutación 773insC en el ex 6 | Pérdida de bipedestación a los 60 meses. Escoliosis |
| 3 | SMARD1 | Nacimiento | Distrés respiratorio | Importantes signos de denervación con fibrilación y potenciales positivos predominio m. distales de extremidades | Cambios polimórficos en el gen IGHMPB2 (ex 1-T57C; ex 6-C714A y G823A; ex 8-C1104T; ex 9-A+18G; ex 13-A2011G y C2316T) | Traqueostomía y ventilación mecánica prolongada. Defunción a los 44 meses de edad |
| 4 | I | 2 meses | Hipotonía | Fibrilaciones y ondas positivas, trazados muy deficitarios de amplitud disminuida, potenciales de unidad motora con aumento de polifásicos | Deleción en heterocigosis del gen SMN1 (deleción de ex 1-6 y deleción de ex 7-8) | Defunción a los 7 meses de edad |
| 5 | I | 3 meses | Hipotonía | Afectación motora con fibrilaciones espontáneas, signos de denervación predominio m. proximales, VC normal | Deleción de una copia del gen SMN1+copia truncada por deleción del ex 1 | Defunción a los 8,5 meses de edad |

En nuestro centro, se realizó el seguimiento de 25 pacientes, 7 de ellos a través de hospitalización a domicilio. La mayoría presentaba AME tipo i (18 casos). El tratamiento de soporte que recibieron incluía rehabilitación y fisioterapia motora (15 pacientes), fisioterapia respiratoria (8 pacientes) o cuidados respiratorios básicos como manejo y aspiración de secreciones (5 pacientes). Dos pacientes recibieron oxigenoterapia domiciliaria mediante cánulas de bajo flujo a los 6 y 7 meses, y en un caso se instauró ventilación no invasiva a la edad de 8 meses. En 2 pacientes se inició ventilación mecánica invasiva y se llevó a cabo traqueostomía. Uno de ellos fue el paciente diagnosticado de SMARD1, en el que se inició soporte ventilatorio invasivo previo a la confirmación diagnóstica. El segundo caso fue un paciente afectado de AME tipo ii, en el que se llevó a cabo una intervención proactiva de forma consensuada con la familia ante la situación de soporte ventilatorio invasivo prolongado, se realizó traqueostomía a la edad de 12 meses y, tras el alta hospitalaria del paciente a los 4 años de edad, se llevó a cabo el seguimiento a través de hospitalización a domicilio hasta su fallecimiento a los 14 años.
Respecto al tratamiento nutricional, 4 pacientes precisaron nutrición enteral domiciliaria a través de sonda nasogástrica y en un caso se realizó gastrostomía (fig. 5). En otro caso, la gastrostomía fue rechazada por los padres.
, ventilación mecánica invasiva (VMI), nutrición enteral mediante sonda nasogástrica (SNG) o gastrostomía.")
Tratamiento de soporte recibido por los pacientes seguidos en nuestro centro. Rehabilitación motora, fisioterapia respiratoria, cuidados respiratorios básicos, oxigenoterapia de bajo flujo, ventilación mecánica no invasiva (VMNI), ventilación mecánica invasiva (VMI), nutrición enteral mediante sonda nasogástrica (SNG) o gastrostomía.
Nos consta el fallecimiento de 18 pacientes del total de los 25 casos seguidos en nuestro centro, siendo la mayoría tipo i (solo un paciente tipo ii ha fallecido en este período). La mediana de la supervivencia en nuestra serie para el tipo i fue de 8,0 meses (rango entre 1,5 y 44 meses) y de 15,8 años para el tipo ii (rango entre 3 y 25,8 años). Destaca que 22/25 de los pacientes afectados de AME tipo i que fueron seguidos fallecieron antes de los 24 meses de edad.
DiscusiónAl final de los 25 años de este estudio, la incidencia para nuestra población de referencia y año es similar a la descrita en otras series, lo cual habla de una estabilidad de penetración genética para esta enfermedad en nuestra población13. La AME tipo i representa el 70,3% de los casos, el defecto genético en la mayoría de los pacientes es la deleción en homocigosis del gen SMN1 (31/36) y la supervivencia media es menor de un año en el tipo i, algo menor a lo descrito en la literatura, y en nuestra serie hay un predominio de varones.
Las manifestaciones clínicas siguen mostrando al lactante hipotónico como el cuadro predominante (fig. 2), si bien en algunos casos (3 en nuestra serie) el comienzo fue la dificultad respiratoria. La edad de inicio de la sintomatología para los AME tipo 1 fue de 2,2 meses. Aunque el diagnóstico fue relativamente temprano, en la actualidad se considera insuficiente para las expectativas que el futuro terapéutico de esta entidad promete14-17. Está en cuestión la utilidad de cribado neonatal para la posibilidad de ofertar a determinados pacientes de inclusión en ensayos clínicos. La técnica ya ha sido implementada y existen estudios en este sentido18.
Entre las familias con pacientes afectados de AME tipo i nos consta una tasa de abortos espontáneos de 19,1%, situada en el límite superior de la cifra descrita en población general (10-20%). No obstante, esta cifra podría infraestimar la tasa real de abortos espontáneos en asociación con la enfermedad, ya que desconocemos la existencia de abortos posteriores en familias sin seguimiento en nuestro centro. Aunque está descrito, son muy escasas las publicaciones sobre la asociación de la AME con abortos espontáneos y no hemos encontrado estudios sistemáticos sobre este tema. El caso publicado relaciona la muerte fetal a la secuencia de hipocinesia-deformación19.
Solo uno de nuestros pacientes presentaba una AME no relacionada con el gen SMN. Fue diagnosticado de un SMARD1. Los polimorfismos hallados en este paciente no han sido descritos hasta ahora. Su asociación al cromosoma 11 junto a la clínica nos llevan a suponer su patogenicidad. El manejo de este caso planteó una situación de soporte ventilatorio precoz mediante traqueostomía al comenzar clínicamente con insuficiencia respiratoria grave sin diagnóstico clínico inicial, presentando el paciente la evolución natural de esta entidad con una afectación ventilatoria grave y en etapas incipientes de la enfermedad, junto a una afectación de predominio distal20. En diversas publicaciones se cuestiona la oportunidad de un soporte respiratorio invasivo para la AME tipo 1 y es un interrogante ético aún por resolver21. Desde 2007 existe un abordaje consensuado de cuidados22 que pretende mejorar la calidad de vida del paciente con AME, si bien su implantación ha sido motivo de controversia según diversos trabajos en distintos países23.
En nuestra experiencia, este nivel de cuidados ha sido puesto en práctica en los últimos años y ha supuesto un esfuerzo de coordinación entre las diversas especialidades implicadas y requiere, además, un adecuado desarrollo de la hospitalización a domicilio para su implementación. Conlleva, como mínimo, la universalización de la oxigenoterapia domiciliaria, la gastrostomía y la ventilación no invasiva en el curso de la enfermedad, además de la optimización de las medidas ortopédicas para evitar deformidades bizarras, siempre de acuerdo con las aspiraciones y expectativas de los padres. Está por dilucidar si este planteamiento alarga la supervivencia y, aunque no es el objetivo de los cuidados, puede tener esta consecuencia lógica. Las asociaciones de pacientes están en la línea de este nivel de cuidados y ello debe ser tenido en cuenta por el pediatra. La supervivencia para los casos tipo i ofreció una mediana de 8 meses, lo cual está en un rango inferior a lo publicado hasta ahora, probablemente por nuestra incorporación tardía al soporte nutricional y respiratorio más activo. Ello no nos permite sacar conclusiones en cuanto a supervivencia global, si bien parece atenuar la ansiedad de los padres en cuanto a la percepción de sufrimiento de su hijo.
En nuestra serie, solo en 2 casos se realizó una traqueostomía. En el caso SMARD1, por lo anteriormente comentado, y en un caso de AME tipo 2, donde la evolución más lenta hace más probable la indicación. El soporte nutricional mediante gastrostomía programada se llevó a cabo en un solo paciente, de acuerdo con los padres, a la edad de 16 meses. El uso de la máquina de tos (cough assistant) se estableció en 2 casos de AME i a los 8 y 23 meses, con desigual satisfacción percibida ya que en uno de los casos empeoró la situación respiratoria y fue abandonada por los cuidadores.
En cuanto a las alteraciones genéticas de nuestra serie, son semejantes a lo previamente publicado (tabla 2). En 2011, las 2 únicas familias en las que se había identificado la copia truncada del gen SMN1 con deleción de exones 1-6 procedían de la misma zona geográfica y en ambos casos dicha copia fue heredada por vía paterna. El análisis de los marcadores polimórficos ligados al gen SMN1 y de los marcadores polimórficos multiloci (272 y 212) mostró que los padres de los 2 pacientes compartían haplotipo, lo que indicaba que ambos cromosomas derivaban de un antepasado común.
Conclusiones y comentariosEn conclusión, nuestra serie ofrece un patrón clínico clásico con diagnóstico relativamente precoz y una incidencia poblacional estable de en torno a un caso por cada 10.000 recién nacidos vivos y año. La mayoría de los casos presenta una genética típica con predominio de varones. En aproximadamente uno de cada 10 pacientes la alteración genética fue diferente de la clásica para el gen SMN. Solo en uno de los 37 pacientes de la serie el gen fue distinto del SMN y correspondió a un SMARD1. La supervivencia de nuestra serie en general ha sido inferior a la descrita previamente. El nivel de cuidados se ha incrementado, en consonancia con las demandas sociales y asistenciales devenidas en los últimos años.
Creemos muy necesario una coordinación multicéntrica para dar una respuesta adecuada al tratamiento y la investigación de esta enfermedad, cuyo futuro es cada vez más esperanzador.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Dra. Carmen de Benito y Dra. Emilia del Castillo, Servicio de Genética, HRU Carlos Haya.
