




Introducción
La agenesia traqueal es una rara malformación congénita del aparato respiratorio que se presenta como dificultad respiratoria severa, cianosis y ausencia de llanto audible al nacimiento1. Es incompatible con una vida prolongada, ya que la mayoría de los pacientes fallecen en las primeras horas o días de vida. Excepcionalmente se han descrito 2 casos de 4 y 6 años de supervivencia2,3. Desde su descripción inicial por Payne4 en 1900, sólo han sido publicados en la literatura especializada alrededor de 150 casos5, con una incidencia aproximada de uno por cada 50.000 recién nacidos vivos. Sin embargo, en España, según los datos del Estudio Colaborativo Español de Malformaciones Congénitas (ECEMC) con un total de 2.310.551 recién nacidos y 36.280 malformados durante un período comprendido de casi 30 años (desde abril de 1976 a diciembre de 2005) no se ha registrado ningún caso de agenesia traqueal. Existe un predominio del sexo masculino (2:1) y antecedente de prematuridad en el 52 % de los casos6,7. La clasificación universalmente aceptada es la que describieron Floyd et al8 en 1962 en la que proponen 3 tipos de agenesia traqueal (fig. 1):
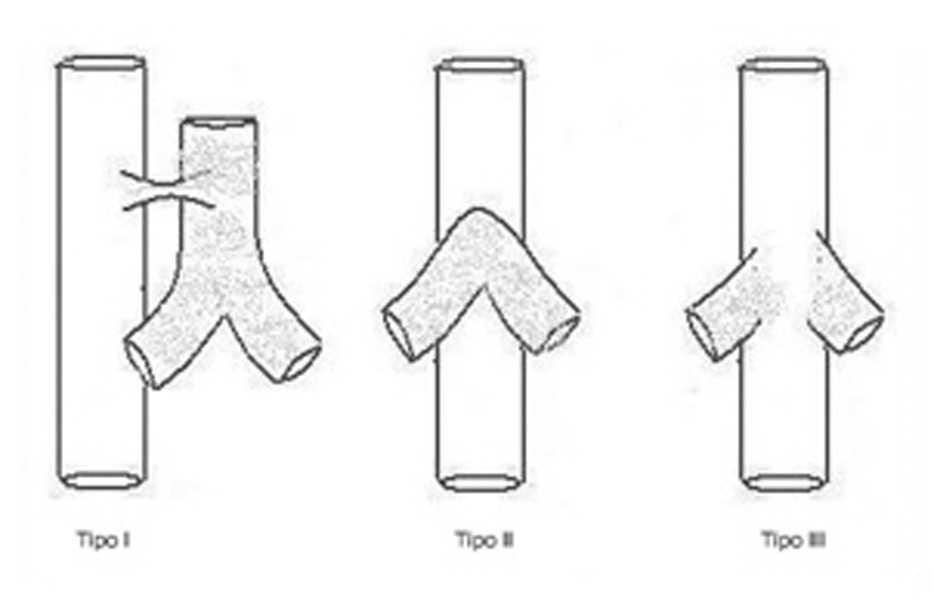
Figura 1. Agenesia traqueal. Clasificación de Floyd.
Tipo I: agenesia de tráquea proximal, con segmento distal de tráquea y bronquios principales normales, acompañándose de fístula traqueoesofágica. Aparece en el 10-15 % de los casos.
Tipo II: agenesia traqueal completa. Bronquios y carina normales, pudiendo existir o no fístula traqueoesofágica. Es el más frecuente (49-59 %).
Tipo III: los bronquios principales nacen independientemente del esófago (30 %).
Otra clasificación alternativa más extensa aunque menos usada fue la descrita posteriormente por Faro et al9 en la que distinguen 7 tipos de agenesia traqueal.
Observación clínica
Madre de 23 años sana, nulípara, sin antecedentes personales de interés, con embarazo según refiere bien controlado en Venezuela hasta el quinto mes de gestación, que acudió a nuestro hospital a las 34 semanas y 5 días de gestación por comenzar con pródromos de parto. En la ecografía prenatal se observó polihidramnios severo, crecimiento fetal menor de lo esperado para su edad gestacional, pie equino, clinodactilia y sospecha de obstrucción intestinal alta. El registro cardiotocográfico era normal. Se realizó cesárea por presentación podálica, prematuridad, polihidramnios severo y dinámica uterina. Se obtuvo un varón con un peso de 1.990 g (P25), una talla de 41 cm (P10) y un perímetro cefálico de 32 cm (P50). Al examen físico externo mostraba pabellones auriculares de implantación baja, micrognatia y retrognatia sin otras alteraciones sugerentes de malformación. Nació cianótico, hipotónico con una frecuencia cardíaca de 120 lat./min, pero con esfuerzos respiratorios que fueron ineficaces a pesar de ventilación con mascarilla facial y presión positiva inspiratoria. Al minuto presentó un test de Apgar de 3. Se realizó laringoscopia directa, se visualizaron cuerdas vocales, y se introdujo un tubo endotraqueal del n.º 3. Mejoró la coloración de piel y mucosas, el tono muscular y sus esfuerzos respiratorios eran aparentemente eficaces por lo que se extubó a los 5 min de vida (test de Apgar 7). Tras su extubación comenzó de nuevo a presentar dificultad respiratoria progresiva con cianosis e imposibilidad de ventilación con mascarilla facial. Se intentó reintubar con ayuda del servicio de anestesia, consiguiendo una aceptable ventilación bilateral, y alcanzando solamente saturación de oxígeno en sangre arterial (SaO2) del 70-75 %. Además precisó administración de adrenalina intravenosa y masaje cardíaco por bradicardia severa. Se trasladó a la unidad de cuidados neonatales y se conectó a ventilación mecánica. Se recolocó el tubo endotraqueal por su excesiva introducción pero no se consiguió aumentar la oxigenación pulmonar a pesar de administrar fracción inspiratoria de oxígeno (FiO2) del 100 %. La radiografía de tórax mostraba un infiltrado difuso en ambos pulmones con broncograma aéreo y falta de expansión. Se decidió su extubación para un nuevo intento pero posteriormente fue imposible la intubación por parte de personal especializado tanto del servicio de pediatría como de anestesia a pesar de sedar y relajar al neonato. Presentaba un importante edema laríngeo y la sospecha de estenosis subglótica era alta considerando ésta la causa de la incapacidad para la intubación. Se intentó ventilación con mascarilla facial y mascarilla laríngea sin éxito y tras presentar parada cardiorrespiratoria falleció a las 2 h y 35 min de vida.
La autopsia realizada reveló los diagnósticos de atresia laríngea subglótica, agenesia traqueal proximal con segmento distal de tráquea y bronquios principales normales (fig. 2), acompañada de fístula traqueoesofágica tipo I (Clasificación de Floyd), hendidura laríngea tipo I (Clasificación de Benjamín e Inglis) (figs. 3 y 4), bilobulación del pulmón derecho, duplicidad ureteral izquierda, hidronefrosis e hidrouréteres derechos, discreto aumento tímico, micrognatia, retrognatia y pabellones auriculares de implantación baja en un varón con peso y talla discretamente disminuidos para su edad gestacional. El cariotipo correspondía a un varón portador de un cromosoma marcador en mosaico "mos 47,XY + mar (43,3)/46,XY (56,6)".

Figura 2. Agenesia traqueal proximal. Segmento distal de tráquea y bronquios principales normales, con fístula traqueoesofágica.
Discusión
Se presenta el caso de un recién nacido con fracaso en el mantenimiento de la vía aérea, que ocasionó gran angustia y frustración por parte del personal médico que le atendió, hasta la confirmación diagnóstica de sus malformaciones en el estudio post mortem.
Nuestro caso forma parte de la agenesia traqueal tipo I de Floyd, el menos frecuente. La ventilación conseguida se mantuvo por la vía esofágica a través de la fístula traqueoesofágica, pese a creer inicialmente que la intubación fue endotraqueal por visualizarse cuerdas vocales. La proximidad del tubo a la fístula permitió la máxima aunque insuficiente ventilación y oxigenación pulmonar como verificamos al recolocar el tubo a su posición correcta.
Durante el embarazo se describe con frecuencia la existencia de polihidramnios (49 %), ocurriendo el nacimiento prematuro en el 64 % de los casos. La presencia de fístula traqueoesofágica se ha encontrado en el 94 % de los casos. En el 90 % de los mismos existe asociación con otras malformaciones congénitas sobre todo cardiovasculares, gastrointestinales y respiratorias7,10,11. Entre las cardiovasculares destacan los defectos septales, defectos valvulares y cardiopatías complejas como transposición de grandes vasos o tetralogía de Fallot así como alteraciones de los vasos umbilicales. Las malformaciones gastrointestinales asociadas son muy amplias, desde anomalías a nivel de esófago, duodeno, estómago, páncreas y bazo hasta imperforación anal y divertículo de Meckel. A nivel respiratorio se incluyen malformaciones laríngeas descritas en el 34 % de los casos como la presencia del cartílago cricoideo elíptico, hipoplasia laringotraqueal o atresia laríngea12,13, y las alteraciones pulmonares (26 %) suelen ser hipoplasia pulmonar o anomalías en la lobulación. Las menos frecuentes se describen a nivel genitourinario (agenesia renal, estenosis uretral), vertebral (hemivértebras, fusiones vertebrales) y de extremidades (hipoplasia de pulgares, agenesia de radio)7. Numerosos autores describieron inicialmente el síndrome VATER que conlleva malformaciones vertebrales, anales, traqueales, esofágicas y renales. Posteriormente la presencia adicional de anomalías cardíacas y de los miembros define la asociación VACTERL. Más recientemente se han descrito la asociación TACRD que incluye atresia/agenesia traqueal, malformaciones cardíacas complejas, defectos radiales y atresia duodenal11. Por ello, la evaluación de las anomalías asociadas debe incluir estudios de imagen como ecografía o escáner y estudios citogenéticos como el cariotipo y/o técnicas de hibridación cromosómica para identificar la presencia de síndromes.
En el caso que nos ocupa existió duplicidad ureteral y bilobulación del pulmón derecho además de la atresia laríngea subglótica y la hendidura laríngea interaritenoidea (figs. 3 y 4). El cariotipo realizado en sangre periférica y en cultivo de piel del recién nacido mostró la existencia de un cromosoma marcador en mosaico, encontrado en el 43,3 % de las células analizadas. Los estudios de hibridación realizados (FISH) descartan a los cromosomas 22 y 15 como cromosomas de origen. Los cariotipos de los padres fueron normales. Como era esperable no se encontró ningún factor teratogénico implicado, y esta alteración cromosómica de novo, producto de anomalías en la meiosis, fue la responsable de las malformaciones halladas. Dado que el riesgo de recurrencia es del 1 %, es importante el diagnóstico prenatal en futuras gestaciones.
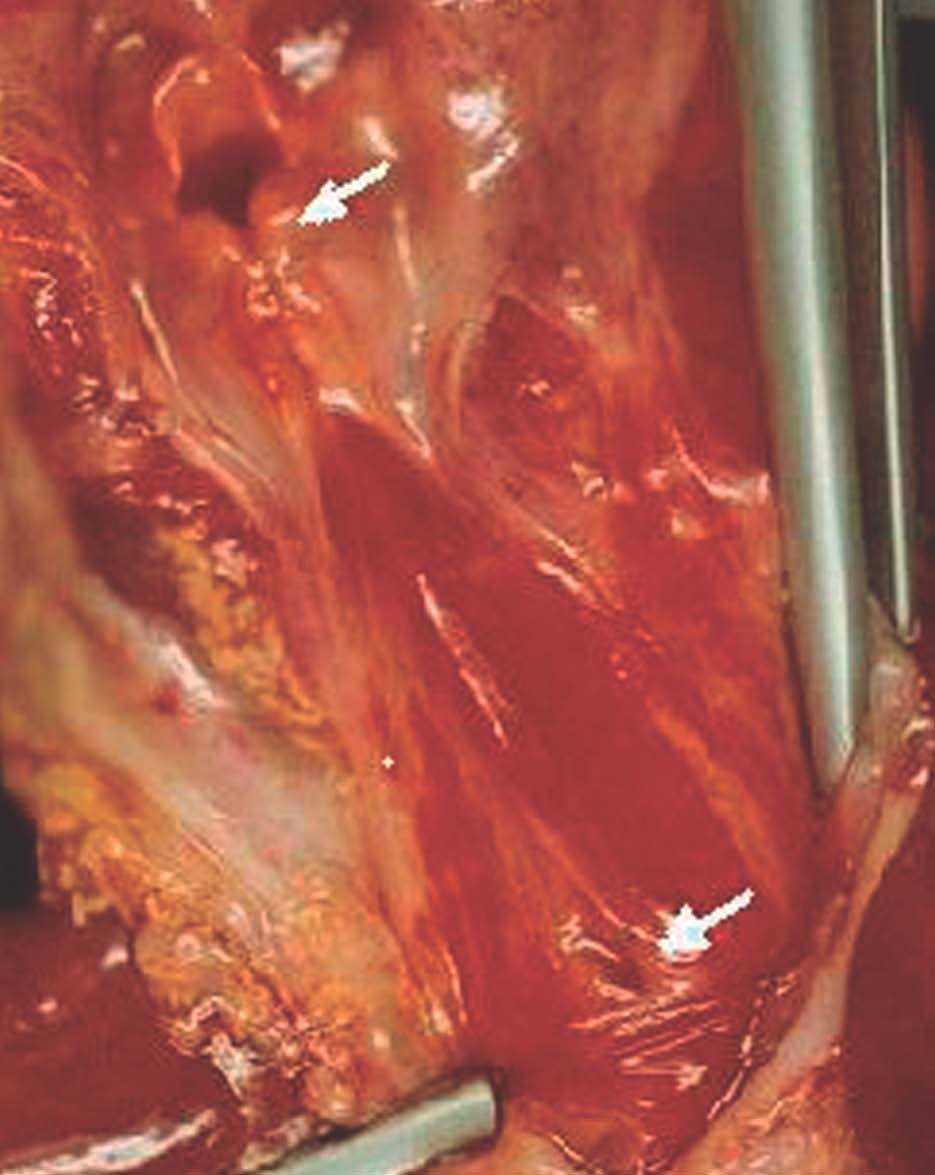
Figura 3. Hendidura laríngea. Fístula traqueoesofágica.
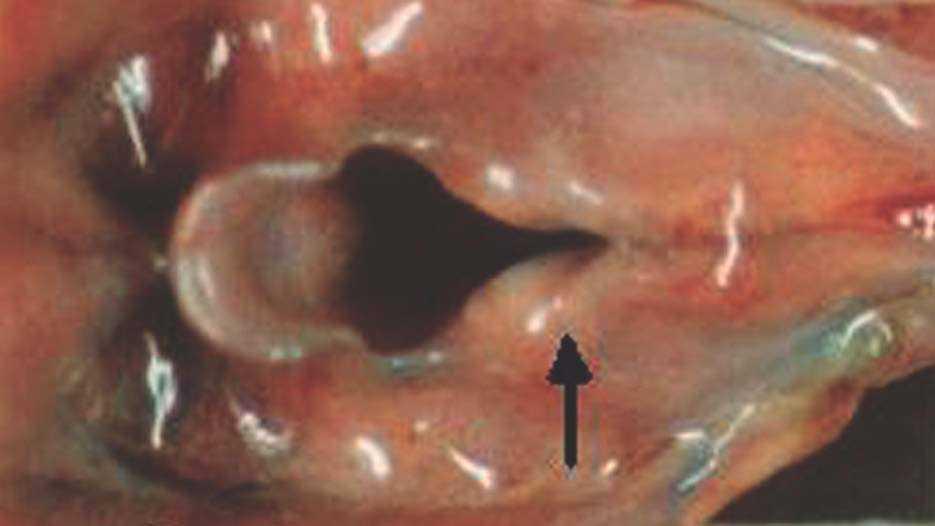
Figura 4. Detalle de la figura anterior. Hendidura laríngea interaritenoidea.
La posibilidad de un tratamiento paliativo depende del tipo de agenesia, ya que el mantenimiento de la vía aérea es esencial para un posible abordaje médico-quirúrgico precoz14. Por ello, ante la sospecha de esta malformación, es necesario poder tener rápido acceso a otras pruebas diagnósticas como la realización de una laringotraqueoscopia o fibrobroncoscopia2,3,15. Si existe fístula traqueoesofágica, la intubación esofágica permitirá la ventilación, como pudimos observar en nuestro caso. Diversas técnicas quirúrgicas como la realización de una esofagostomía superior para derivar la saliva y secreciones, una inferior para intubación y mantenimiento de la ventilación, además de incluir una gastrostomía y una ligadura esofágica inferior como medida antirreflujo, han permitido aumentar la supervivencia en un caso hasta los 10 meses de edad y en dos hasta los 4 y 6 años de edad, respectivamente2,3,15. Sin embargo, el problema fundamental en estos casos es la reconstrucción esofágica aunque se ha intentado con interposición de colon o estómago3. La oxigenación por membrana extracorpórea (ECMO) podría utilizarse inmediatamente al nacimiento en aquellos casos con alta sospecha prenatal mientras se evalúa la posibilidad de una terapia quirúrgica.
Se han empleado diversas técnicas para la reconstrucción definitiva de la vía aérea desde prótesis sintéticas de marlex, silastic y dacron en animales16 hasta injertos homólogos de pericardio o de vejiga sin obtener ningún resultado exitoso17,18. La reconstrucción traqueal con cartílago costal podría ofrecer una buena solución a largo plazo19. Los aloinjertos de tráquea han mostrado resultados favorables en el tratamiento de la estenosis traqueal en pacientes pediátricos, pero todavía no se han utilizado en la agenesia traqueal completa20.
En cualquier caso, los posibles injertos en un futuro tendrán que permitir un crecimiento y desarrollo normales, ser capaces de mantener la vía aérea limpia de secreciones y de resistir cambios de presiones. En el tratamiento de estas malformaciones hay que tener en cuenta las anomalías asociadas, sus posibles opciones quirúrgicas así como las complicaciones añadidas. Hoy en día no existe una terapia definitiva eficaz que garantice una buena calidad de vida a largo plazo.
Correspondencia: Dra. M. Rupérez Lucas.
Servicio de Neonatología. Fundación Hospital Alcorcón.
Avda. Budapest, s/n. 28922 Alcorcón. Madrid. España.
Correo electrónico: martaruperez@telefonica.net
Recibido en abril de 2006.
Aceptado para su publicación en marzo de 2007.
