










La hipertransaminasemia es un hallazgo frecuente en pediatría, puede ser banal o reflejar enfermedad grave potencialmente tratable. El objetivo de este documento es establecer, mediante la revisión de la evidencia disponible, un consenso para un adecuado enfoque práctico desde la detección de la hipertransaminasemia hasta completar su estudio en la edad pediátrica. Para ello, se constituyó un grupo de trabajo con participación de miembros de la Sociedad de Gastroenterología, Hepatología y Nutrición Pediátrica (SEGHNP), Asociación Española de Pediatría de Atención Primaria (AEPap) y Sociedad Española de Pediatría de Atención Primaria (SEPEAP). Se establecieron 21 recomendaciones con el objetivo de que sirvan de utilidad en la práctica clínica habitual tanto en atención primaria como hospitalaria.
Hypertransaminasemia is a frequent finding in pediatrics, which could reflect potentially treatable serious disease. The aim of this document is to establish, by reviewing the available evidence, a consensus for an adequate management of hypertransaminasemia, from its detection until the study is complete. To this end, a working group was formed with the participation of members of the Society of Pediatric Gastroenterology, Hepatology and Nutrition (SEGHNP), the Spanish Association of Primary Care Pediatrics (AEPap) and the Spanish Society of Primary Care Pediatrics (SEPEAP). Twenty-one recommendations are established with a marked practical component that will be useful in hospital clinical practice and primary care.
Las transaminasas son enzimas intracelulares presentes en los hepatocitos y otras células. Las más importantes son la glutámico-oxalacético transaminasa (GOT) o aspartato aminotransferasa (AST), menos específica debido a su ubicuidad y la glutámico-pirúvico transaminasa (GPT) o alanina aminotransferasa (ALT), más específica hepática1–5.
La hipertransaminasemia puede aparecer en el contexto del estudio de una enfermedad hepática o de forma casual en un estudio analítico realizado por otras causas6. Elevaciones leves de transaminasas generan con frecuencia evaluaciones incompletas7. Una hipertransaminasemia persistente necesita una evaluación sistemática para detectar etiologías concretas6–8.
El objetivo de este documento es establecer mediante el consenso de pediatras expertos en atención primaria y en gastroenterología, hepatología y nutrición pediátrica, los pasos a seguir desde la primera analítica con hipertransaminasemia (HTRA) hasta completar el proceso diagnóstico. Se incluye la secuencia de pruebas complementarias a realizar (fig. 1). No se ha contemplado en la elaboración de este documento el estudio del paciente con colestasis aislada o asociada a hipertransaminasemia, el proceso diagnóstico de cada patología específica una vez sospechada, el tratamiento de las diferentes causas de hipertransaminasemia ni el manejo de la disfunción hepática o del fallo hepático agudo (FHA).
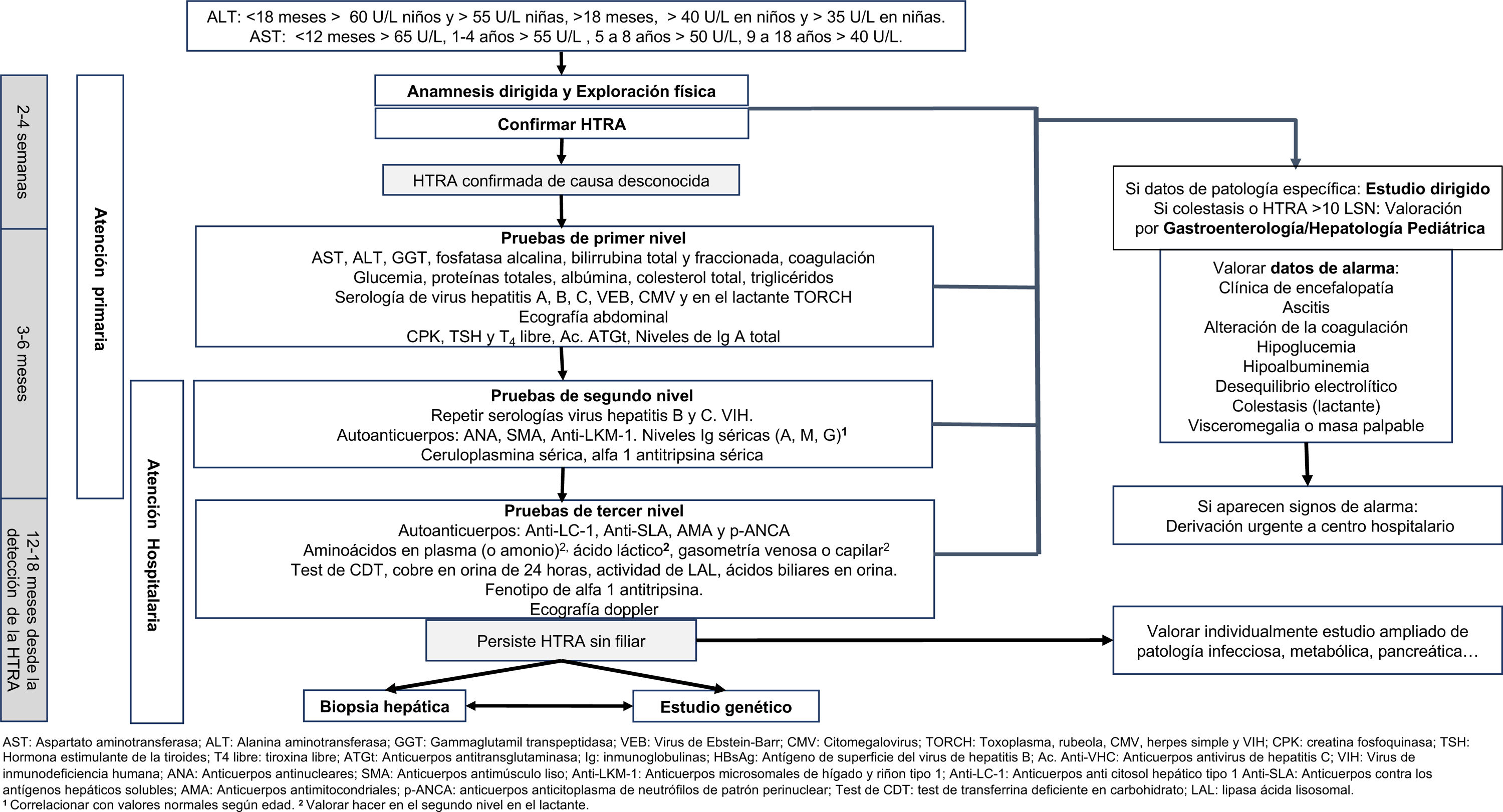
Algoritmo diagnóstico. Ac. Anti-VHC: anticuerpos antivirus de hepatitis C; ALT: alanina aminotransferasa; AMA: anticuerpos antimitocondriales; ANA: anticuerpos antinucleares; Anti-LC-1: anticuerpos anti citosol hepático tipo 1; Anti-LKM-1: anticuerpos microsomales de hígado y riñon tipo 1; Anti-SLA: anticuerpos contra los antígenos hepáticos solubles; AST: aspartato aminotransferasa; ATGt: anticuerpos antitransglutaminasa; CMV: citomegalovirus; CPK: creatina fosfoquinasa; GGT: gammaglutamil transpeptidasa; HBsAg: antígeno de superficie del virus de hepatitis B; Ig: inmunoglobulinas; LAL: lipasa ácida lisosomal; p-ANCA: anticuerpos anticitoplasma de neutrófilos de patrón perinuclear; VEB: virus de Epstein-Barr; TORCH: toxoplasma, rubeola, CMV, herpes simple y VIH; SMA: anticuerpos antimúsculo liso; Test de CDT: test de transferrina deficiente en carbohidrato; TSH: hormona estimulante de la tiroides; T4 libre: tiroxina libre; VIH: virus de inmunodeficiencia humana.
aCorrelacionar con valores normales según edad.
bValorar hacer en el segundo nivel en el lactante.
Se estableció un grupo de trabajo (GT) con 8 miembros, representando a la SEGHNP, AEPap y SEPEAP. Se plantearon 15 preguntas clínicas sobre qué hacer, cuándo y cómo, desde la detección inicial de la HTRA hasta completar el estudio tanto en atención primaria como hospitalaria.
Se realizó una búsqueda bibliográfica sistemática en Pubmed, que incluía desde enero 2010 a enero 2020, con búsqueda «liver test» «liver disease» de 0-18 años, «aminotransferase» «hipertransaminasemia» de 0-18 años sin límite temporal y «genetic liver disease» «histology liver disease» para cualquier edad y sin límite temporal. Se hizo una segunda búsqueda desde enero 2020 a septiembre 2020 siguiendo la misma metodología que en la primera. Se empleó el gestor bibliográfico compartido Zotero 5.0. Se incluyeron 248 artículos en inglés y castellano, de los que se obtuvo el artículo completo. De ellos, 113 se consideraron relevantes para este consenso y fueron seleccionados por adecuarse a las preguntas planteadas en la elaboración de esta guía.
Elaboración del documentoLas preguntas clínicas fueron respondidas (cada pregunta fue contestada por 2 miembros del grupo) en base a la evidencia disponible y con el objetivo de establecer un estudio diagnóstico ordenado de la hipertransaminasemia basado en la opinión del GT. No se ha realizado una revisión sistemática ni un metaanálisis de las pruebas de estudio. Se realizaron 5 revisiones completas (Google Drive®) por todos los miembros del GT, con reunión virtual (GoToMeeting® cedida por la SEGHNP y Zoom®) tras cada revisión. Todas las recomendaciones que aparecen en el documento han sido consensuadas y aceptadas por los miembros del grupo (consenso informal). El documento de consenso completo puede consultarse en las páginas web de cada una de las sociedades participantes.
Definición de hipertransaminasemia y causas principalesEn ausencia de acrónimo aceptado para hipertransaminasemia, se consensuó emplear HTRA para el documento.
La HTRA representa la elevación de los niveles séricos de transaminasas por encima del límite superior de normalidad (LSN)9 de una muestra sana y representativa de la población estudiada. Estos niveles presentan una considerable variabilidad biológica intra e interindividual. Su elevación indica lisis de las células que las contienen o aumento de la permeabilidad de sus membranas2,5,6. En la población pediátrica, por extrapolación del límite clásico en adultos, se ha utilizado habitualmente >40U/l, pero los referidos por distintos autores10–19 son relativamente diferentes y variables entre ellos (tabla 1). Hay un considerable entrecruzamiento entre poblaciones sanas y enfermas, lo que dificulta la obtención de puntos de corte con sensibilidad y especificidad apropiadas9. Además, algunos autores señalan que, en la adolescencia, los valores habituales son demasiados altos y pierden sensibilidad cuando se utiliza como cribado inicial para la detección de esteatosis hepática o hepatitis crónica por VHC, por lo que deberemos fijar un punto de corte de normalidad más bajo en estas circunstancias10.
Valores pediátricos de transaminasas según diferentes autores
| Autor (año) | Valores en U/l, según rango de edad y/o sexo | |||
|---|---|---|---|---|
| Fraser (2007) | Adolescentes, chicos y chicas | |||
| ALT LSN=30 | ||||
| England (2009) | <18 meses, niños | <18 meses, niñas | >18 meses, chicos | >18 meses, chicas |
| ALT LSN=60 | ALT LSN=55 | ALT LSN=40 | ALT LSN=35 | |
| Lai (2009) | ALT p2,5-p97,5=8-38 | |||
| Schwimmer (2010) | Adolescentes chicos | Adolescentes chicas | ||
| ALT LSN=26 | ALT LSN=22 | |||
| Rodoo (2013) | 6 meses-8 años, niños | 6 meses-8 años, niñas | 9-18 años, chicos | 9-18 años, chicas |
| ALT LSN=23,4 | ALT LSN=17,4 | ALT LSN=30,6 | ALT LSN=30,6 | |
| 6-12 meses, niños y niñas | 1-4 años, niños y niñas | 5-8 años, niños y niñas | 9-18 años, chicos y chicas | |
| AST LSN=66 | AST LSN=55,8 | AST LSN=48 | AST LSN=43,2 | |
| Zierk (2015) | ALT y AST expresados en intervalos continuos de referencia, como gráfica de percentiles 2,5 a 97,5, en función de la edad | |||
| Klietherme (2017) | Adolescentes chicos | Adolescentes chicas | ||
| ALT LSN=28 | ALT LSN=21-24 | |||
| Bussler (2018) | 11 meses-16 años, chicosVarios rangos de edad | 11 meses-16 años, chicasVarios rangos de edad | ||
| ALT LSN chicos=29,9-38 | ALT LSN chicas=24,2-31,7 | |||
| AST LSN chicos=41,5-68,7 | AST LSN chicas=35,2-62,9 | |||
| Kim (2018) | Adolescentes chicos | Adolescentes chicas | ||
| ALT LSN=33 | ALT LSN=25 | |||
| Liu (2019) | 9 rangos de edad, diferencia sexo solo en adolescentes | |||
| ALT LSN=39-56 | ||||
| AST LSN=32-67 | ||||
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; LSN: límite superior de la normalidad; p: percentil
No existe correlación fiable entre el grado de elevación de las transaminasas y la gravedad o el pronóstico. Sin embargo, las cifras muy elevadas suelen asociar daño extenso por lo que la HTRA se divide arbitrariamente en 3 grados: leve (<5×LSN), moderada (=5-10×LSN) y muy elevada (>10×LSN). Por otra parte, de forma empírica y por consenso tradicional, la HTRA se define como crónica cuando se prolonga más de 6 meses6.
Toda HTRA, aunque en ocasiones transitoria, debe considerarse como marcador potencial de enfermedad y ser interpretada en el contexto clínico del paciente. En consecuencia, debe ser confirmada siempre y seguida hasta su normalización o diagnóstico etiológico1,20.
Las causas de HTRA en pediatría son múltiples, tanto hepáticas como extrahepáticas, por lo que el diagnóstico diferencial es amplio (tabla 2)8,20. La elevación aislada o desproporcionada de AST suele ser debida a causas extrahepáticas: extracción dificultosa, hemólisis, rabdomiólisis, etc. También puede ser debida a la formación de macro-AST3.
Causas de hipertransaminasemia en pediatría
| Hepáticas | Hepáticas | ||
|---|---|---|---|
| Infecciosa | Hepatitis agudas (A, B, C, D y E) y crónicas (B y C) Sistémicas con tropismo hepático: VEB, CMV, VHS, VVZ, VIH, toxoplasma, SARS-CoV-2. Sepsis y bacteriemias. Brucelosis, leptospirosis, fiebre tifoidea. Otras (más en niños pequeños): virus respiratorios y gastrointestinales (adenovirus, VRS, parvovirus, echovirus, rotavirus), gastroenteritis por Salmonella, ITU | Neoplásicas | Primarias: tumores hepáticos (hepatoblastoma, hepatocarcinoma) Secundarias: neuroblastoma, linfomas, leucemias, metástasis |
| Hepatobiliares | Colelitiasis, quiste de colédocoCursan con patrón colestásico, precisan listado y estudio aparte | ||
| Traumática | Trauma obstétrico (hematoma subcapsular), trauma abdominal | ||
| Miscelánea | Cromosomopatías (síndrome de Turner), amiloidosis, sarcoidosis, síndrome de Reye, APLV (en lactantes), enfermedades vasculares | ||
| Tóxicas | Fármacos, drogas, tóxico, sustancias de herbolario y medicina alternativa | ||
| Inmunitarias | Hepatitis autoinmune, colangitis autoinmune. Hepatopatía asociada a enfermedades inmunitarias: colagenosis y otras | Enfermedad digestiva o sistémica | Enfermedad celíaca, disfunción tiroidea, fibrosis quística, enfermedad inflamatoria intestinal crónica, síndrome de Shwachman-Diamond |
| Obesidad | Hígado graso no alcohólico (esteatosis hepática, esteatohepatitis) | Extrahepáticas | |
| Metabólicas | Déficit de alfa-1-antitripsina, enfermedad de Wilson, galactosemia, fructosemia, tirosinemia, glucogenosis (I, III, IV, VI y IX), lipidosis (Gaucher, Nieman-Pick), trastornos beta-oxidación ácidos grasos, déficit de lipasa ácida lisosomal, trastornos congénitos de la glicosilación, defectos de la síntesis de ácidos biliares, defectos del ciclo de la urea, alteración del metabolismo de las lipoproteínas (HBL), porfirias, otras | Aumento de AST y ALT | Enfermedades musculares y neuromusculares (AST>ALT): distrofias de Duchenne y Becker, caveolinopatías, distrofias de miembros y cintura, polimiositis, dermatomiositis, miopatías metabólicas (glucogenosis tipo V o enfermedad de McArdle). Otras causas de afectación muscular: grandes traumatismos, quemaduras extensas, cirugía, ejercicio intenso. Alteraciones tiroideas: hipotiroidismo e hipertiroidismo. Nefropatías. Insuficiencia suprarrenal. Anorexia nerviosa |
| Isquémicas-vasculares | Bajo gasto cardiaco, obstrucción arterial hepática o venosa portal, insuficiencia cardíaca congestiva, síndrome de Budd-Chiari | Aumento aislado de AST | Hemolisis: Extracción dificultosa de la muestra, enfermedades hemolíticas. Enfermedades cardiacas (pericarditis, miocarditis, infarto agudo de miocardio). Macrotransaminasemia aislada |
ALT: alanina aminotransferasa; APLV: alergia proteínas de la leche de vaca; AST: aspartato aminotransferasa; CMV: citomegalovirus; HBL: hipobetalipoproteinemia; ITU: infección del tracto urinario; VEB: virus Epstein-Barr; VHS: virus herpes simple; VIH: virus inmunodeficiencia humana; VRS: virus respiratorio sincitial; VVZ: virus varicela-zóster.
Aparecen en negrita las etiologías más frecuentes.
La etiología más frecuente de elevación aguda leve-moderada y con normalización en los 6 primeros meses, son las infecciones víricas, respiratorias o gastrointestinales en lactantes y en niños más mayores la infección por citomegalovirus y virus de Epstein-Barr8. También hay que pensar en un posible origen farmacológico o tóxico. Dentro de las causas farmacológicas, las más frecuentes son los antibióticos y antiepilépticos, pero existe una gran variedad de fármacos que potencialmente pueden producir HTRA. Otras posibles causas tóxicas son las sustancias de abuso (por ej. alcohol, cocaína), disolventes, pesticidas o sustancias de herbolario (p. ej., hierba de San Juan, efedra o genciana).
La principal causa de HTRA persistente de origen hepático en niños mayores y adolescentes es el hígado graso no alcohólico (conocido por su acrónimo en inglés, NALFD) relacionado con sobrepeso/ obesidad8.
Recomendaciones- 1.
Recomendamos estratificados por edad y sexo, los siguientes puntos de corte de ALT para población sana europea: para menores de 18 meses >60U/l en niños y >55U/l en niñas, y para mayores de 18 meses >40U/l en niños y >35U/l en niñas. Para el cribado inicial de esteatosis hepática o hepatitis crónica por VHC en el adolescente, se proponen los siguientes valores de ALT: 26U/l en niños y 22U/l en niñas.
- 2.
Proponemos estratificados por edad, los siguientes puntos de corte de AST: >65U/l para menores de un año, >55U/l de 1 a 4 años, >50U/l de 5 a 8 años y >40U/l de 9 a 18 años.
- 3.
La HTRA puede ser de origen hepático y/o extrahepático. Se recomienda una búsqueda activa escalonada en función de la edad y las causas más frecuentes, tanto hepáticas como extrahepáticas.
La anamnesis y la exploración física son las primeras herramientas diagnósticas ante una HTRA5,21,22. En ocasiones existen datos que informan sobre la etiología o la gravedad del proceso (tabla 3)5,20–22. Los antecedentes personales y familiares, consumo de sustancias o fármacos, realización de ejercicio intenso, extracciones analíticas previas o síntomas sistémicos deben ser recogidos20,22. La exploración será detallada por aparatos ya que existe una amplia variedad de enfermedades extrahepáticas que pueden acompañarse de HTRA6 (tabla 2).
Síntomas y signos en la anamnesis y exploración física
| Manifestaciones inespecíficas | Astenia, anorexia, náuseas, vómitos, distensión abdominal, dolor abdominal |
| Datos de afectación hepática | Hepatomegalia, ictericia, acolia, coluria |
| Manifestaciones de hepatopatía crónica | Telangiectasias, eritema palmar, malnutrición, ascitis, esplenomegalia, circulación colateral, hemorragia digestiva |
| Rasgos dismórficos | Fenotipo característico (síndrome de Alagille) |
| Manifestaciones neurológicas | Irritabilidad, alteración del carácter, alteración del tono muscular, retraso psicomotor |
| Manifestaciones de afectación muscular | Alteraciones de la marcha, dolor muscular, hipertrofia o debilidad muscular |
A nivel analítico, el primer paso es confirmar la HTRA4. El perfil hepático a nivel analítico comprende marcadores de lesión hepatocelular, de colestasis y de síntesis hepática (tabla 4)5,20. La ecografía abdominal es útil para valorar la morfología hepática y de las vías biliares, descartar procesos tumorales, y en el estudio de adolescentes con sobrepeso u obesidad, para orientar hacia la presencia de un hígado graso. El estudio serológico viral se orientará según los antecedentes del paciente6.

Muchas de las HTRA son autolimitadas, pero existe variabilidad en cuanto al tiempo de resolución (semanas o meses)6,22. No hay evidencia sobre el tiempo necesario para repetir el análisis. Clásicamente se recomienda de 2 a 4 semanas ante HTRA sin datos de alarma5,20–22. Este intervalo deberá individualizarse en situaciones temporales de HTRA (toma de fármacos o infusiones herbales hepatotóxicas, infecciones, ejercicio intenso…) cuya resolución puede requerir más tiempo6,20.
La presencia de datos de alarma (tabla 5) requiere la derivación urgente a un centro hospitalario. De especial relevancia será descartar el FHA (incapacidad del hígado de cumplir sus funciones de biosíntesis, regulación y detoxificación)23. En cuanto aparezcan signos de encefalopatía, disfunción renal tributaria de diálisis, inestabilidad hemodinámica o compromiso respiratorio, el tratamiento debe realizarse en unidades de cuidados intensivos, y valorar, en presencia de encefalopatía, hipoglucemia, insuficiencia renal o INR>2,524, el traslado a un hospital con capacidad para realizar trasplante hepático.
Recomendaciones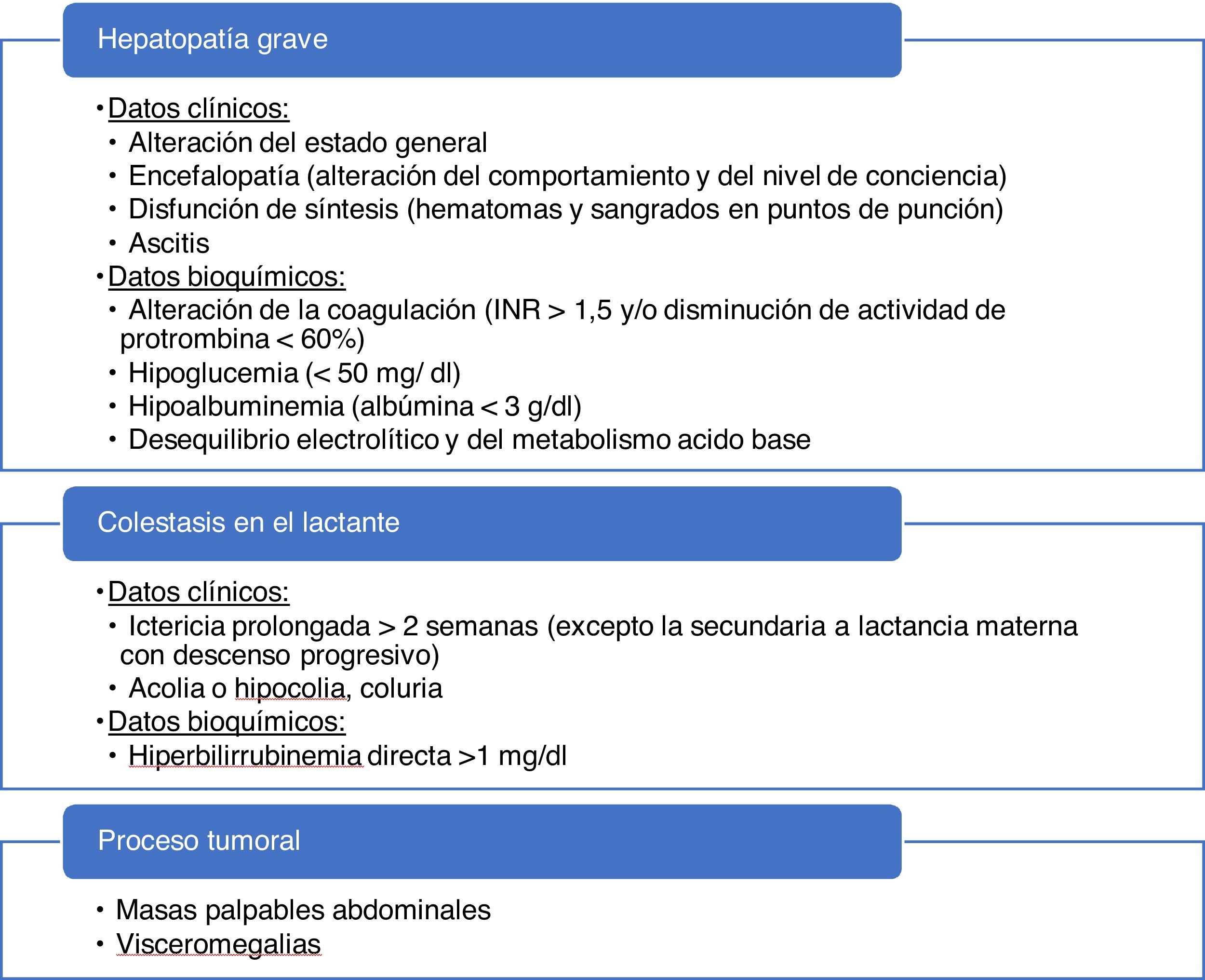
- 1.
Proponemos realizar una adecuada anamnesis y exploración física que nos permitirá, en ocasiones, orientar la sospecha diagnóstica y valorar la presencia de manifestaciones de gravedad.
- 2.
Recomendamos que toda HTRA, aunque en ocasiones sea transitoria, se considere como factor potencial de patología, se sitúe en el contexto clínico del paciente y se evalúe inicialmente con un perfil hepático completo (tabla 4).
- 3.
Toda HTRA debe ser confirmada. Sugerimos confirmar la HTRA, en ausencia de datos de alarma, dentro de un periodo de 2 a 4 semanas desde su detección. Si existe la sospecha de una causa temporal específica, podemos demorar el control hasta incluso 8 semanas, si con ello esperamos resolver ese proceso transitorio. Siempre que existan datos clínicos y/o analíticos de gravedad el control se realizará de forma urgente.
- 4.
Tras su confirmación, o coincidiendo con esta, deben valorarse inicialmente las causas más frecuentes o relevantes de HTRA mediante análisis de sangre y estudio ecográfico (pruebas de primer nivel) (tabla 6).
Tabla 6.Pruebas de primer, segundo y tercer nivel
Primer nivel Perfil hepático completo AST, ALT, GGT, fosfatasa alcalina, bilirrubina total y fraccionada, estudio de coagulación Bioquímica general Glucemia, proteínas totales, albúmina, colesterol total, triglicéridos, CPK Enfermedad infecciosa aguda Serología viral orientada de hepatitis A, B, C, virus de Epstein-Barr, citomegalovirus, y en el lactante TORCH Prueba de imagen Ecografía abdominal Otras Función tiroidea (TSH y T4 libre), celiaquía (anticuerpos anti-transglutaminasa de tipo IgA), niveles de IgA total Segundo nivel Enfermedad infecciosa Repetir serologías hepatitis B, C y VIH Autoanticuerpos ANA, SMA, anti-LKM-1 Otras Ceruloplasmina sérica, alfa-1-antitripsina sérica, Ig séricas (IgA, IgG e IgM)a Tercer nivel Autoanticuerpos Anti-LC-1, anti-SLA, AMA, p-ANCA Prueba de imagen Ecografía doppler abdominal Otras Aminoácidos en plasma (o amonio)b, gasometría venosa o capilarb, ácido lácticob, test de CDT, fenotipo de alfa-1-antitripsina, determinación de cobre (orina de 24 h), actividad de LAL, ácidos biliares (plasma y/o orina), ALT: alanina aminotransferasa; AMA: anticuerpos antimitocondriales; ANA: anticuerpos antinucleares; anti-LC-1: anticuerpos anticitosol hepático tipo 1;; anti-LKM-1: anticuerpos microsomales de hígado y riñon tipo 1; anti-SLA: anticuerpos contra los antígenos hepáticos solubles; AST: aspartato aminotransferasa; CPK: creatin fosfoquinasa; GGT: gammaglutamil transpeptidasa; Ig: inmunoglobulinas; LAL: lipasa ácida lisosomal; p-ANCA: anticuerpos anticitoplasma de neutrófilos de patrón perinuclear; SMA: anticuerpos antimúsculo liso; test de CDT: test de transferrina deficiente en carbohidrato; TORCH: toxoplasma, rubeola, citomegalovirus, herpes simple y VIH; TSH: hormona estimulante de la tiroides; T4 libre: tiroxina libre; VIH: virus de la inmunodeficiencia humana.
- 5.
Recomendamos evaluar en todas las visitas los datos de alarma que indiquen la necesidad de una derivación urgente a un centro hospitalario (hepatopatía grave, colestasis del lactante o sospecha de proceso tumoral). La colestasis del niño (no lactante) deberá ser valorada de forma preferente.
- 6.
En caso de sospecha de FHA, recomendamos valorar ingreso en una unidad de cuidados intensivos y traslado a un centro con programa activo de trasplante hepático pediátrico.
Ante la ausencia de signos o síntomas que permitan orientar las pruebas diagnósticas, es fundamental continuar con un estudio ordenado y sistemático que debe incluir determinadas enfermedades metabólicas25–28.
Es importante considerar que la NAFLD es, actualmente, la principal causa de HTRA en el niño mayor de 10 años. La presencia de obesidad y/o hiperecogenicidad hepática en la ecografía inicial nos pueden orientar, pero no son diagnósticas de NAFLD29 y requieren evaluación de acuerdo con su protocolo específico29.
La realización de ecografía con doppler permite el diagnóstico de patologías que podrían cursar con alteraciones en la vascularización hepática30 y es útil además para hacer una valoración de la presencia de hipertensión portal31.
El estudio de otras enfermedades que, debido a su menor frecuencia o porque la HTRA aislada no es su principal manifestación, se realizará de manera individualizada y dirigida en función de la edad, el contexto, la posible sintomatología y hallazgos clínicos asociados (tabla 7). Las pruebas de primer, segundo y tercer nivel se recogen en la tabla 6.
Entidades a evaluar de manera individual
| Entidades | Características | Pruebas |
|---|---|---|
| Errores innatos del metabolismoSu diagnóstico se realizará guiado por especialista en enfermedades metabólicas | Muchas de ellas están incluidas en los programas de cribado neonatal.Es muy poco frecuente que cursen asintomáticas y sus principales manifestaciones (fallo de medro, talla baja, vómitos, retraso psicomotor, hepatomegalia, hipoglucemia colestasis, etc.) suelen aparecer en periodo de lactante | Revisar cribado neonatalRequieren estudio de aminoácidos en sangre, amonio, lactato, metabolitos acumulados en sangre y/u orina, estudios enzimáticos específicos y estudio genético |
| InfeccionesSu diagnóstico se realizará guiado por especialista en enfermedades infecciosas | Grupos de riesgo de VHB o VHC con cribado inicial negativo o paciente inmunocomprometido | PCR de ADN de VHB y PCR de ARN de VHC.Infecciones oportunistas |
| Viajero | Parásitos y bacterias poco frecuentes en nuestro medio | |
| Afectación pancreática | Fibrosis quística | Revisar cribado neonatalCloro en sudor/estudio genético gen CFTR |
| Síndrome de Shwachman-Diamond | CitopeniasEstudio genético | |
| MacroAST | Elevación aislada, sostenida y exclusiva de AST por presencia de macro enzimas | Porcentaje de AST precipitada con polietilenglicol (%PPA)Electroforesis |
ADN: ácido desoxirribonucleico; ARN: ácido ribonucleico; AST: aspartato aminotransferasa; PCR: proteína C reactiva; VHB: virus de hepatitis B; VHC: virus de hepatitis C.
- 1.
Tras las pruebas de primer nivel, si persiste la HTRA no filiada, sugerimos estudio analítico de hepatitis autoinmune, enfermedad de Wilson, déficit de alfa-1-antitripsina, déficit de lipasa ácida lisosomal, defectos de la síntesis de ácidos biliares, trastornos congénitos de glicosilación, defectos del ciclo de la urea y repetir serologías de virus de hepatitis B (VHB) y C (VHC).
- 2.
En el niño con ecografía compatible con esteatosis hepática y /o sobrepeso se recomienda seguir el protocolo específico de estas enfermedades para completar el estudio de HTRA.
- 3.
Ante una HTRA persistente sugerimos completar el estudio radiológico con ecografía doppler hepática para valorar la vascularización venosa y arterial del hígado.
- 4.
Si existen datos compatibles con enfermedad metabólica y en función del tipo de cribado neonatal, de la clínica, la edad del paciente y las alteraciones analíticas, se indica realizar un estudio dirigido con la participación de un especialista en enfermedades metabólicas. En caso de sospecha de fibrosis quística se recomienda realizar test de cloro en sudor y, en función del resultado, estudio genético del gen CFTR.
- 5.
En niños pertenecientes a grupos de riesgo de VHB o VHC (inmunodeprimidos, pacientes procedentes de áreas con alta prevalencia e hijos de padres o madres portadores de VHB o VHC) a pesar de antígeno HBs o anticuerpos anti-VHC negativos, proponemos realizar PCR de ADN de VHB y PCR de ARN de VHC.
- 6.
En determinadas situaciones epidemiológicas, especialmente en el niño inmigrante, en el paciente con sospecha o inmunosupresión confirmada y como parte del estudio de una afectación sistémica, se debe ampliar la búsqueda de patología infecciosa en colaboración con un especialista en enfermedades infecciosas.
- 7.
Ante la elevación aislada y mantenida de AST indicamos valorar la presencia de macro-AST.
No existen trabajos específicos que avalen los tiempos de realización de controles en la HTRA. El contexto del paciente es muy importante para determinar la frecuencia de su realización. Una única determinación normal no descarta un problema hepático ya que algunas enfermedades hepáticas pueden presentar un curso fluctuante6. Además, algunas enfermedades pueden presentar elevaciones leves de transaminasas y producir un FHA6,32.
El motivo de derivación a un centro hospitalario, en ausencia de datos de alarma (tabla 5), será para completar el proceso diagnóstico-terapéutico, una vez superadas las posibilidades de atención primaria y ante datos de posible gravedad (HTRA>10 veces LSN, colestasis, etc.)20
Recomendaciones- 1.
Se aconseja realizar un control analítico a los 3-6 meses a los pacientes con HTRA inexplicada y resuelta inicialmente, evitando cerca de la extracción situaciones que puedan producir aumento de transaminasas secundarias (infección, medicación, ejercicio, etc.)
- 2.
En el paciente asintomático con HTRA, en ausencia de datos de alarma, proponemos la realización del estudio de primer y segundo nivel en los primeros 3-6 meses. Si persiste HTRA no filiada y asintomática, sugerimos realizar estudio completo (primer, segundo y tercer nivel) antes de 12 meses tras la detección de la HTRA.
- 3.
Los pacientes con colestasis asociada o HTRA muy elevadas (>10 veces LSN) recomendamos que sean valorados de manera preferente por parte de un pediatra gastroenterólogo/hepatólogo.
- 4.
En ausencia de datos de alarma, indicamos derivar a gastroenterología/hepatología pediátrica a los pacientes con HTRA con sospecha etiológica concreta que requieran diagnóstico o tratamientos específicos y a aquellos con HTRA sin filiar que requieran pruebas de tercer nivel. El lugar de realización de las pruebas de segundo nivel dependerá de la organización, experiencia y posibilidades de cada área.
Las pruebas complementarias previas, el fenotipo del paciente y el análisis del caso con el genetista, nos ayudarán en la elección del tipo de estudio genético a realizar. Las técnicas de secuenciación de nueva generación (New Generation Secuenquencing [NGS]) son las técnicas empleadas en la actualidad. Nos permiten detectar mutaciones puntuales, pequeñas pérdidas y ganancias intragénicas en varios genes simultáneamente. Los estudios que se emplean en la clínica con NGS son: panel de genes diana33, exoma clínico (EC) (lectura del exoma que puede dirigirse a través de las características fenotípicas principales del paciente mediante el uso de nomenclatura de HPOs (Human Phenotype Ontology)34 y exoma completo (lectura de todo el exoma genético)35. Debido al avance de las técnicas de genética molecular y su mayor disponibilidad, la realización de este estudio será cada vez más habitual y precoz en la práctica clínica.
Biopsia hepáticaDebido a que es un método intervencionista, se realiza después de una evaluación inicial exhaustiva y no invasiva. El método de elección es la punción guiada con ecografía utilizando agujas de diferente calibre en función de la edad, con técnicas de aspiración o de corte36. La HTRA mantenida es la principal causa de su realización en niños37. El momento de realizarla no está tan claramente establecido. Algunos autores han descrito un bajo rendimiento diagnóstico de la biopsia hepática (BH) ante HTRA mantenida sin sospecha etiológica y esta podría posponerse hasta los 14-16 meses de evolución8. Los padres o tutores deben ser informados del riesgo beneficio en función de la naturaleza de la enfermedad hepática de su hijo.
La BH nos dará información sobre las características histopatológicas específicas de diferentes enfermedades hepáticas38,39. Además, es útil como herramienta para valorar el pronóstico de enfermedades hepáticas crónicas mediante índices de actividad histológica (inflamación, fibrosis, necrosis, esteatosis, etc.). En el estudio anatomopatológico se pueden usar diversas técnicas: tinción básica con hematoxilina-eosina, técnicas histoquímicas específicas de tinción, técnicas inmunohistoquímicas o la utilización de microscopía electrónica37. La comunicación entre el equipo de anatomía patológica y el clínico es crucial para el adecuado procesamiento y valoración de la muestra histológica.
En los últimos años se han desarrollado técnicas no invasivas de valoración de fibrosis hepática mediante elastografía. Aunque aporta información interesante para el seguimiento, no contamos con valores de normalidad validados en niños ni valores específicos para las diferentes enfermedades pediátricas por lo que hasta la fecha no sustituyen a la realización de la BH40.
Recomendaciones- 1.
En el contexto de un paciente con HTRA mantenida sin diagnóstico etiológico, deberá realizarse un estudio genético con la colaboración del equipo de genetistas clínicos. En este tipo de pacientes, en los que la HTRA es el único signo guía, recomendamos la realización de un exoma clínico.
- 2.
Sugerimos realizar BH (preferiblemente guiada por ecografía) entre los 12 y 18 meses de evolución de una HTRA asintomática mantenida, sin otros signos de alarma, con estudio etiológico previo no diagnóstico.
Los autores de este consenso no presentan ningún conflicto de intereses.
