






Vascular anomalies are changes in vascularization that usually appear in the foetal stage, at birth or in early childhood. They can cause chronic pain, motor impairment, cosmetic changes or coagulopathy and may be fatal in some cases, but in every case they have a negative impact on the quality of life of the child and the family. Up to 150 different subtypes have been described. They can involve arteries, capillaries, veins, lymphatic vessels or a combination thereof. They may be associated with additional malformations and frequently cause musculoskeletal and soft tissue hypertrophy or hypotrophy. They can develop anywhere in the body, invade any tissue and affect the function of various organs. The prevalence of the different subtypes varies greatly, from 1/20 to 1/1 000 000. Subtypes considered rare diseases (incidence <1/2000) continue to affect more than 500 000 people in the European Union.
Differentiating between vascular tumours and vascular malformations is critical, especially in paediatric patients. They are completely different diseases, although they are often grouped under the umbrella term of vascular anomalies.
The diagnostic algorithm used in the clinical evaluation of vascular anomalies should be based on a thorough history-taking and detailed physical examination.
Future knowledge in this field will be based above all on genetic findings and therapeutic innovations. New molecules and their indications are being explored with the aim of reducing the aggressiveness of previous treatments and increasing the life expectancy and quality of life of patients who do not respond to conventional treatments.
Las anomalías vasculares son alteraciones de la vascularización que suelen aparecer en el feto, al nacimiento o en la primera infancia. Pueden causar dolor crónico, disfunción motora, trastornos cosméticos, coagulopatía, y son ocasionalmente mortales, pero en todos los casos empeoran la calidad de vida del niño y su familia. Existen hasta 150 subtipos diferentes descritos. Pueden afectar arterias, capilares, venas, linfáticos o a una combinación de ellos. Pueden estar asociadas a malformaciones adicionales y causan con frecuencia hipertrofia o hipotrofia musculo-esquelética y de partes blandas. Pueden localizarse en cualquier parte del cuerpo, invadir cualquier tejido y afectar a la función de diversos órganos. Las prevalencias de los diferentes subtipos varían enormemente, de 1/20 a 1/1.000.000. Los subtipos considerados enfermedades raras (incidencia inferior a 1/2000) siguen afectando a más de 500.000 personas en la UE.
La diferenciación entre tumores vasculares y malformaciones vasculares es crítica, especialmente cuando se trata de pacientes pediátricos. Las dos entidades representan patologías completamente diferentes, aunque agrupadas bajo el paraguas común del término Anomalías Vasculares.
El algoritmo diagnóstico utilizado en la evaluación clínica de las anomalías vasculares debe basarse en una historia clínica completa y un examen físico detallado
El futuro del conocimiento en este campo se basará sobre todo en los hallazgos genéticos y en las innovaciones terapéuticas. Nuevas moléculas y sus indicaciones están siendo exploradas, con destino a reducir la agresividad de tratamientos previos y de dar esperanza y calidad de vida a los pacientes sin respuesta a los tratamientos convencionales.
Vascular anomalies are changes in vascularization that usually appear in the foetal stage, at birth or in early childhood. They can cause chronic pain, motor impairment, cosmetic changes or coagulopathy and may be fatal in some cases, but in every case they have a negative impact on the quality of life of the child and the family. Up to 150 different subtypes have been described. They can involve arteries, capillaries, veins, lymphatic vessels or a combination thereof. They may be associated with additional malformations and frequently cause musculoskeletal and soft tissue hypertrophy or hypotrophy. They can develop anywhere in the body, invade any tissue and affect the function of various organs. The prevalence of the different subtypes varies widely, from 1/20 to 1/1 000 000. Subtypes considered rare diseases (incidence <1/2000) continue to affect more than 500 000 individuals in the European Union (Figs. 1 and 2).


There is a long history of attempts to diagnose and classify AVs that started with the indiscriminate use of eponyms and descriptive terms such as “port wine stain”, “strawberry birthmark” or “cavernous angioma”, among others.
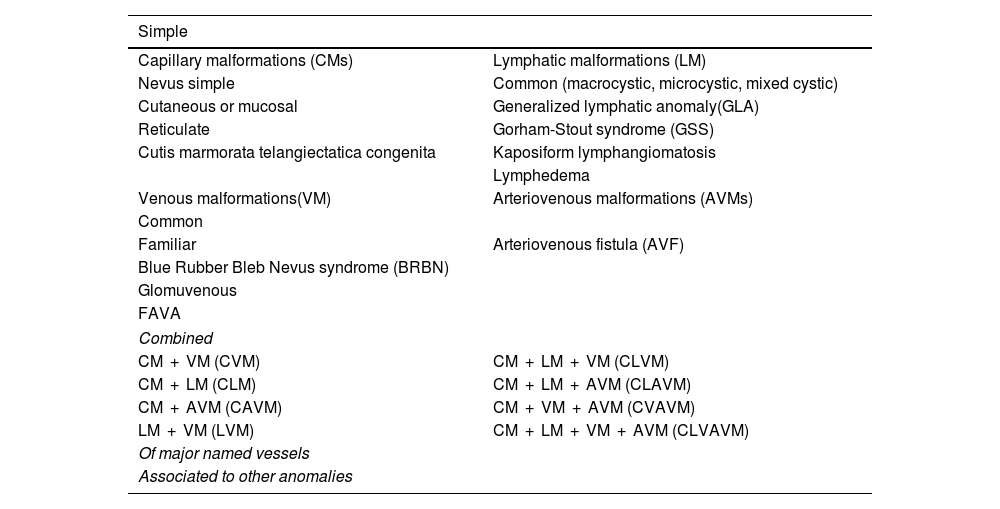
The rapid proliferation of this rather unscientific nomenclature brought attention to the need to develop a biological classification of VAs. One such classification was introduced by John Mulliken in 1982,1 setting the foundations for the subsequent classification of the International Society for the Study of Vascular Anomalies (ISSVA2,3 (Tables 1 and 2).
Vascular tumours.
| Benign | Locally aggressive | Malignant |
|---|---|---|
| Infantile haemangioma | Kaposiform haemangioendothelioma | Angiosarcoma |
| Congenital haemangioma | Retiform haemangioendothelioma | Epithelioid haemangioendothelioma |
| Rapidly involuting (RICH) | Papillary intralymphatic angioendothelioma | |
| Non-involuting (NICH) | Dabska tumour | |
| Partially involuting (PICH) | Composite haemangioendothelioma | |
| Tufted angioma | Pseudomyogenic haemangioendothelioma | |
| Spindle-cell haemangioma | Polymorphous haemangioendothelioma | |
| Epithelioid haemangioma | Haemangioendothelioma not otherwise specified | |
| Pyogenic granuloma | Sarcoma de Kaposi | |
| Other |
Vascular malformations.
| Simple | |
|---|---|
| Capillary malformations (CMs) | Lymphatic malformations (LM) |
| Nevus simple | Common (macrocystic, microcystic, mixed cystic) |
| Cutaneous or mucosal | Generalized lymphatic anomaly(GLA) |
| Reticulate | Gorham-Stout syndrome (GSS) |
| Cutis marmorata telangiectatica congenita | Kaposiform lymphangiomatosis |
| Lymphedema | |
| Venous malformations(VM) | Arteriovenous malformations (AVMs) |
| Common | |
| Familiar | Arteriovenous fistula (AVF) |
| Blue Rubber Bleb Nevus syndrome (BRBN) | |
| Glomuvenous | |
| FAVA | |
| Combined | |
| CM + VM (CVM) | CM + LM + VM (CLVM) |
| CM + LM (CLM) | CM + LM + AVM (CLAVM) |
| CM + AVM (CAVM) | CM + VM + AVM (CVAVM) |
| LM + VM (LVM) | CM + LM + VM + AVM (CLVAVM) |
| Of major named vessels | |
| Associated to other anomalies | |
VAs, arteriovenous; C, capillary; CLVAV, capillary-lymphatic-venous-arteriovenous malformation; M, malformation; V, venous.
Future knowledge in the field will be chiefly based on genetic discoveries and therapeutic innovations. Nearly 50 genetic variants have already been identified in different Vas, and the application of whole genome sequencing (WES) to affected tissues will expand this catalogue exponentially (Figs. 3–5).



Age is considered a key factor in the clinical assessment of VAs, as they may manifest with different signs and symptoms in the foetus, infant, child and adult in close association with the stage of life.
The diagnostic algorithm used in the clinical evaluation of VAs must be based on a thorough history-taking and detailed physical examination. The most important aspects to explore in the anamnesis are whether the lesion was present at birth (vascular malformations) and its growth rate (proportional or disproportional to the child’s growth). The exception is found in congenital haemangiomas: rapidly involuting congenital haemangioma (RICH) and non-involuting congenital haemangioma (NICH). Both proliferate exclusively in the foetal period.
Differentiating between haemangioma and congenital vascular malformations is critical, especially in paediatric patients. They are completely different diseases, although they are grouped under the umbrella term of vascular anomalies. Malformations are errors of vascular morphogenesis in the context of somatic mosaicism and, more rarely, germline mutations. Vascular tumours may be benign (the majority), locally aggressive or malignant (angiosarcoma and epithelioid haemangioendothelioma).
Vascular tumoursInfantile haemangiomaHaemangiomas of infancy, or infantile haemangiomas (HIs) are the most common type of paediatric vascular tumours and have a specific aetiology, presentation, prognosis and treatment. These lesions are proliferative and usually are not present at birth, developing in the first weeks of life, and have a self-limiting course with rapid proliferation and slow regression. Most have undergone atrophy and full involution, with mild residual telangiectasia and a residual mass of fibrofatty tissue, by age 10 years. Histologically, they are positive for glucose transporter-1 (GLUT-1) on staining through every stage of growth and involution, which facilitates their diagnosis, especially in deep extracutaneous tumours.
Congenital haemangiomaInfantile haemangiomas must be distinguished from congenital haemangiomas, as their treatment is different. Congenital haemangiomas develop fully in utero and do not undergo postnatal proliferation. Histologically, they are negative for GLUT-1.
In the group of congenital haemangiomas, 3 types or phenotypes have been described: RICH, NICH and partially involuting congenital haemangioma (PICH). Rapidly involuting congenital haemangiomas involute spontaneously within the first year of life, while NICHs persist for life.
Another important difference between infantile and congenital haemangiomas is that the former are not caused by any known mutations, whereas the latter are caused by GNA spectrum variants (GNAQ/GNA11).
Infantile haemangioma associated with a syndromeApproximately 30% of patients with facial segmental infantile haemangiomas may develop a syndrome with posterior fossa malformations or other structural lesions in the central nervous system (SNC), segmental haemangioma, anomalies in the major arteries, heart and eyes and deformities of the sternum or midline of the trunk (PHACES).4
The evaluation must include magnetic resonance imaging of the head, neck and trunk, an ophthalmological evaluation, cardiac ultrasound and thyroid function tests.
Symptomatic airway haemangiomas are associated with a segmental mandibular distribution of the cutaneous haemangioma.5
Segmental haemangiomas in the midline of the lower back may be associated with congenital anomalies of the kidneys, sacrum, spine or genitourinary system in what is known as LUMBAR syndrome or PELVIS syndrome.6
Treatment of haemangiomaWhen it comes to treatment, steroid therapy and surgery were the standard of care for complicated IH for half a century. In 2008, cases of haemangioma involution were described in patients treated with oral propranolol, a nonselective beta-blocker. Propranolol became the first-line treatment for IH, and its efficacy and safety were confirmed in a multicentre, randomised double-blind trial that confirmed that a 6-month course of propranolol at a dose of 3 mg/kg/day was efficacious for treatment of IH with minimal adverse effects.7
Since then, numerous studies have been conducted to determine which patients need treatment and the appropriate timing and duration of treatment. In 2016, a consensus guideline on the management of IH was published in Spain that established the following indications for treatment with propranolol: potentially lethal IH, IH causing functional impairment, ulcerated or painful IH, IH not responding to basic wound care and IH associated with a risk of facial or body disfigurement requiring future reconstructive surgery. The guideline also recommends initiating treatment at the earliest opportunity, with a duration of 6 months, with clinical monitoring and dose adjustment based on body weight, although some patients may require prolongation of treatment beyond 12 months.8
Laser or pulsed light therapy can attenuate the superficial coloration of residual telangiectasias. Combined with propranolol, it is also useful for treatment of ulcerated haemangiomas, alleviating pain and promoting healing of the ulcer from the first session.9
The management of congenital haemangiomas is conservative for the first few years (except in cases of liver involvement with heart failure),10 but is essentially surgical later on in the management of sequelae, after involution or in lesions associated with significant functional impairment or cosmetic defects.11,12
Congenital haemangiomas do not respond to propranolol or any other pharmacological agents known to date, but the identification of the involved gene variant can be a first step in the management of non-involuting CH through the use of specific inhibitors.13
Other vascular malformationsKaposiform haemangioendothelioma (KHE) and tufted angioma are two tumours that have a close association from a histological and clinical perspective, with a characteristic lymphatic infiltrate and the development, when they reach a large size, of severe thrombocytopenia and coagulopathy (Kassabach-Merritt phenomenon).14
The first case of successful treatment of a severe case of KHE with rapamycin was reported in 2010.15 Since then, numerous publications have confirmed the effectiveness of this treatment, which achieves clinical improvement, recovery of the platelet count and correction of coagulopathy with minimal side effects, so it has since been established as the first-line treatment.16–19
Vascular malformationsVascular malformations are structural anomalies of the circulatory system, as opposed to tumours. These lesions nearly always result from abnormal vascular embryogenesis due to a mutation and are present at birth, although they are not always visible. The natural history of these anomalies is heterogeneous, and in rare cases they may spontaneously regress. They are classified chiefly based on the type of vessel involved as:
- •
Arteriovenous malformations (AVMs)
- •
Venous malformations (VMs)
- •
Lymphatic malformations (LMs)
- •
Capillary malformations (CMs)
Arteriovenous malformations are high-flow structural anomalies with abnormal arteriovenous communications. In newborns, they may appear as a soft pink lesion that cannot be easily differentiated from a CM. Lesions in deeper tissues may go undetected for years. Their progression depends on many factors, including the causative gene (MAP2K1, KRAS, RASA1, BRAF, PTEN, among others) ranging from initial quiescence to heart failure or life-threatening bleeding in the advanced stages.
Arteriovenous malformations in the extremities may manifest with hypertrophy of the bone and soft tissue of the affected extremity.20
Venous malformationsVenous malformations are anomalies in venous embryogenesis that are nearly always caused by changes in TEK-TIE2 and less frequently in other genes such as PIK3CA, the glomulin gene or MAP3K3.21
These malformations may be classified as:
- none-
Extratruncular or spongiform VMs: they can be found in most tissues, usually in the skin or muscle, in the form of networks of venous lakes connected through microvenules to the deep venous system.22
- none-
Truncular VMs (of major named vessels): these lesions manifest as segmental dilatations or aberrant aneurysms (persistent sciatic vein or lateral marginal vein).23
Venous malformations are associated with intravascular coagulopathy caused by the sluggish blood flow within, giving rise to thrombophlebitis, D-dimer elevation, formation of phleboliths and chronic disabling pain.24 In addition, these lesions tend to keep growing,25 partly due to the local coagulopathy.26–28
Lymphatic malformationsThe nomenclature of LMs continues to be unclear, and generic terms such as lymphangioma, lymphangiectasia or lymphatic dysplasia are already outdated to describe the multiple associated malformations.21 At present, LMs are classified based on their clinical presentation, morphology and extension as: common LM, generalised lymphatic anomaly (with bone and visceral involvement), central conducting lymphatic anomaly (chylothorax, chylous ascites, chylopericardium), kaposiform lymphangiomatosis, LM in Gorham-Stout disease and linfedemas.28,29
The vast majority are caused by changes in the PIK3CA gene, and a small group by changes in the KRAS and HRAS genes.30
Capillary malformationsCapillary malformations are the most frequent type of VA and manifest as congenital dilatation of the superficial dermal vessels of the skin present at birth that grow proportionally to the child. They remain for life and do not tend to involute. The natural history of CMs is variable and depends on the anatomical location of the lesion.
Capillary malformations may be associated to other vascular malformations and when they present in specific locations they are considered pathognomonic:
- none-
CM involving face and/or eyes, leptomeningeal vascular malformation and glaucoma: Sturge-Weber syndrome.
- none-
CM in upper lip and nostrils, megalencephaly and polymicrogyria: macrocephaly-capillary malformation syndrome.
- none-
CM of the lower lip, lymphatic malformation predominant on the face and neck, asymmetry, and partial/generalized overgrowth (CLAPO syndrome).
- none-
CM in midline of the back and medullary AVM: Cobb syndrome.
- none-
CM in fingertips and lymphatic-venous malformation in the upper extremity: finger and nails capillary anomaly.
- none-
CM in lateral trunk or thighs and lymphatic-venous malformation with overgrowth: PIK3CA-related overgrowth spectrum (PROS)
Traditionally, the treatment of VAs consisted in pressotherapy, laser, embolization and radical or palliative surgery. These treatments achieved partial control of symptoms but could not halt progression and, in many instances, did not improve the quality of life of patients.31
The true breakthrough in the management of vascular anomalies in the past 10 years was the development of targeted treatment for inhibition of the mutations present in each tumour or malformation subtype.32 Sirolimus or rapamycin is an mTOR (mammalian target of Rapamicyn) protein inhibitor that, before its use in VAs, was chiefly used to prevent allograft rejection in kidney transplant recipients. In addition to its immunomodulatory activity, sirolimus has antiangiogenic and antiproliferative effects that make it a more than appropriate tool for the management of symptomatic VAs.33 Initially it was tried in compassionate use for management of anomalies refractory to other treatments, but the good outcomes achieved, especially in patients with lymphatic malformations, expanded its indications and it is currently used, even as first-line treatment, in multitude conditions. In common LMs, sirolimus reduces the size of the lesion, eliminates lymph leakage and reduces the number of cysts and the frequency of intralesional bleeding episodes and infection. In complex LMs, sirolimus stabilises osteolysis, halting its progression, and reduces the severity of pericardial or pleural effusion and ascites. In addition, as we already mentioned in relation to KHE, sirolimus can also correct coagulopathy. Although it is usually administered orally at a dose of 0.8 mg/m2 every 12 h, there is also evidence of improvement of superficial microcystic LMs with topical administration of sirolimus.34
The effectiveness and safety of treatment with sirolimus has been confirmed, as it has proven to be harmless with minimal adverse effects, even in neonates. However, the optimal dose, serum drug concentration and duration of treatment have yet to be established. The current evidence suggests that treatment with sirolimus should be individualised and that doses and target drug concentrations should be tailored in each patient based on the phenotype and clinical manifestations.35
To alleviate pain associated with thrombi or phleboliths, aspirin may be given at a dose of 50 mg/day, although there is insufficient evidence on its efficacy.36 If this is not effective, the administration of low molecular weight heparin via the subcutaneous route is effective from the first dose, with a recommended treatment duration of at least 2 weeks.37 Oral factor Xa inhibitors such as rivaroxaban, approved for use in children, or factor IIa inhibitors, such as dabigatran, are anticoagulants whose use is increasing in these patients, allowing for prolonged treatment in very severe cases, and their combination with rapamycin seems to be effective in reducing the size of these lesions.38–40
The discovery of PIK3CA variants in patients with VAs has led to the incipient use of PIK3CA inhibitors, initially developed for treatment of cancer, in these patients, with promising results.
Alpelisib is an oral inhibitor of the p110α subunit of class I phosphatidylinositol 3-kinase (PI3KCA) in the 2-aminothiazole derivative group. In vitro, alpelisib exhibits potent inhibition of the p110α subunit of PI3K with a good safety profile. Alpelisib had only been authorised for use in combination with fulvestrant for PIK3CA-mutated, hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2−) advanced or metastatic breast cancer, but the United States Food and Drug Administration (FDA) recently authorised its use in patients with congenital VAs carrying this variant. In 2018, Canaud described a murine model of VAs with overgrowth similar to the disease in humans.41,42 The model demonstrated the efficacy of alpelisib in preventing and improving organ dysfunction. The article also described the compassionate use of alpelisib in 19 patients (adult and paediatric) with PROS, which achieved improvement of symptoms in every patient. The treatment achieved a decrease in size in previously intractable vascular tumours, with improvement of congestive heart failure, reduction of hemihypertrophy and attenuation of scoliosis. The treatment was not associated with any significant adverse events. This case series was the first to provide direct evidence in support of the use of PIK3CA inhibitors as a promising treatment option for PROS.
At present, treatment with alpelisib in patients with PROS is provided through expanded access and in the framework of 4 clinical trials. Since there is already a significant number of patients in treatment with alpelisib for this rare indication, a retrospective review is being conducted to analyse clinical and functional outcomes and the safety profile of this treatment.
The data available to date indicate that 75% of patients respond to treatment and 25% show a very significant reduction in lipomatous masses, and no patient has dropped out of treatment due to serious adverse events.42
As we already discussed, changes in KRAS, NRAS, BRAF and MAP2K1 have been identified in patients with arteriovenous and lymphatic malformations that may yield targeted treatments in the future. MEK1 inhibitors, which are already used in cancer and neurofibromatosis type 1, may be tried in compassionate use in these patients, for whom few non-invasive treatment options are currently available. Trametinib and, more recently, selumetinib have already been established as treatment options in patients with high-flow malformations, malformations with osteolysis and kaposiform lymphangiomatosis caused by KRAS variants.43
New molecules and indications are being investigated with the aim of reducing the aggressiveness of previous treatments and to give hope and improve quality of life in patients who do not respond to conventional treatment.
Conflicts of interestThe authors have no conflicts of interest to declare.
