





Birth asphyxia is one of the principal causes of early neonatal death. In survivors it may evolve to hypoxic-ischaemic encephalopathy and major long-term neurological morbidity. Prolonged and intense asphyxia will lead to energy exhaustion in tissues exclusively dependent on aerobic metabolism, such as the central nervous system. Energy deficit leads to ATP-dependent pumps blockage, with the subsequent loss of neuronal transmembrane potential. The most sensitive areas of the brain will die due to necrosis. In more resistant areas, neuronal hyper-excitability, massive entrance of ionic calcium, activation of NO-synthase, free radical generation, and alteration in mitochondrial metabolism will lead to a secondary energy failure and programmed neuronal death by means of the activation of the caspase pathways. A third phase has recently been described that includes persistent inflammation and epigenetic changes that would lead to a blockage of oligodendrocyte maturation, alteration of neurogenesis, axonal maturation, and synaptogenesis. In this scenario, oxidative stress plays a critical role causing direct damage to the central nervous system and activating metabolic cascades leading to apoptosis and inflammation. Moderate whole body hypothermia to preserve energy stores and to reduce the formation of oxygen reactive species attenuates the mechanisms that lead to the amplification of cerebral damage upon resuscitation. The combination of hypothermia with coadjuvant therapies may contribute to improve the prognosis.
La asfixia intraparto es una de las causas más frecuentes de muerte neonatal precoz pero también puede, en los supervivientes, evolucionar a una encefalopatía hipóxico-isquémica responsable de una elevada morbilidad neurológica. La presencia de episodios de hipoxia-isquemia prolongados conduce a un rápido agotamiento energético en los tejidos exclusivamente dependientes del metabolismo aeróbico, como el sistema nervioso central. El déficit energético conlleva una paralización de las bombas ATP-dependientes y subsiguiente pérdida del potencial neuronal transmembrana. La población neuronal de las regiones más sensibles del SNC mueren por necrosis, mientras que en otras áreas se produce una hiperexcitabilidad neuronal con entrada masiva de calcio iónico, activación de NO-sintasa, generación de radicales libres que alteran el funcionamiento mitocondrial, provocando un fallo energético secundario y muerte neuronal por apoptosis. Recientemente se ha propuesto una tercera fase en la que factores como la inflamación persistente y los cambios epigenéticos causarían un bloqueo de la maduración de los oligodendrocitos, alteración de la neurogénesis, del crecimiento axonal y de la sinaptogénesis. En este contexto, el estrés oxidativo va a tener un papel protagonista como responsable tanto en causar daño directo al SNC como en activar cascadas metabólicas conducentes a la apoptosis e inflamación. La hipotermia moderada precoz, al preservar las reservas energéticas y disminuir la formación de especies reactivas de oxígeno, atenuará el daño cerebral posreanimación. La combinación de la hipotermia con terapias coadyuvantes para modular el estrés oxidativo podría contribuir a mejorar el pronóstico.
So-called oxy-regulator tissues, such as the central nervous system (CNS) and myocardium, require high amounts of energy to maintain membrane potentials and therefore depend on aerobic metabolism, which generates energy much more efficiently than anaerobic metabolism.1 In the CNS, the transmission of action potentials involves ATP-dependent ion pumps, which use a large amount of energy. Furthermore, the CNS cannot store energy in forms that can be mobilised rapidly, such as phosphocreatine or glycogen, and therefore depends on a continuous supply of glucose and oxygen. In consequence, oxygen and glucose deprivation lead to a rapid depletion of energy stores and cell death in a matter of minutes.2–4
Intrapartum asphyxia is characterised by periods of hypoxia/ischaemia during labour that, depending on severity, may result in death or cause hypoxic-ischaemic encephalopathy (HIE). The mortality due to HIE is of 1 to 8 deaths per 1000 live births in developed countries and can be as high as 26 deaths per 1000 live births in developing countries. The main challenge for neonatologists is to reduce the morbidity and mortality associated with HIE, since despite the widespread use of hypothermia, as many as 45% of these patients still die today, while a high percentage of survivors develop significant disabilities.5
The purpose of this review article is to describe the biochemical basis of oxidative metabolism as well as the adjuvant therapies that may help control the production of free radicals and improve the outcomes of hypothermia.
Oxidative metabolism2,6–9Aerobic metabolism and oxidative phosphorylation (Fig. 1)In multicellular organisms, oxygen must be available for mitochondrial oxidative phosphorylation to produce the energy required to sustain life. Substrates such as glucose, amino acids and fatty acids are converted to acetyl coenzyme A (acetyl-coA) in the mitochondrial matrix. Acetyl-coA is metabolised by the different components of the Krebs cycle. This process releases high-energy electrons that are carried to the electron transport chain by nicotinamide adenine dinucleotide and flavin adenine dinucleotide. The electron transport chain consists of a series of adjacent enzyme complexes (i, iii and iv) capable of pumping protons (H+) to the intermembrane space, thus generating a membrane potential (ψm). ATP-synthase then allows protons to flow back into the matrix down the electrochemical gradient, which releases energy used to synthesise ATP from ADP. Oxygen accepts the electrons produced by the Krebs cycle, preventing damage to mitochondrial structures. Energy is used so efficiently that 36 ATP molecules are obtained from 1 glucose molecule and 106 ATP molecules from 1 palmitic acid molecule.2,6–9
Reactive oxygen species and free radicals (Fig. 2)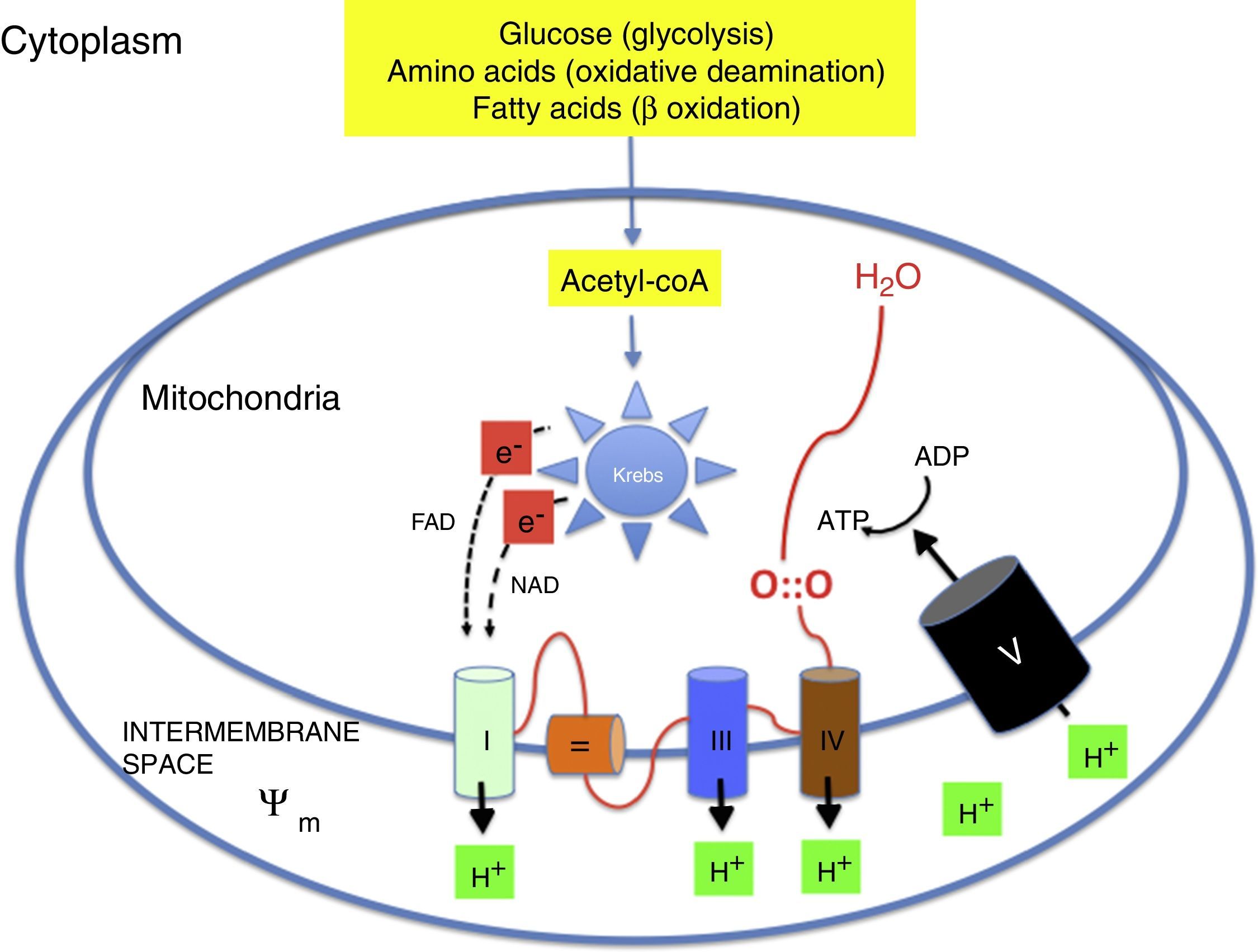
Oxygen is reduced by the addition of 4 electrons to its outermost shell. Oxygen has a limited reactivity, so it is reduced sequentially (slow reduction). However, its reactivity increases exponentially in the presence of transition metals like copper or iron (rapid reduction). The abundant presence of iron in the CNS thus increases the speed of oxidative processes, as occurs in HIE. In the sequential process, oxygen is reduced by a single electron to a superoxide anion (O2−), by two electrons to hydrogen peroxide (H2O2), or by three electrons to a hydroxyl radical (OH). All these substances, which are common products of mitochondrial oxidative phosphorylation, are known as reactive oxygen species. However, some of these species, like the superoxide anion, the hydroxyl radical and others like nitric oxide (NO) or lipid peroxy radicals (LOO) are also free radicals, that is, species with a single unpaired electron in their outer shell that are therefore highly reactive and interact with nearby substrates to obtain the additional electron they require to attain stability, generating a chain reaction that can damage cellular structures. The most prevalent free radical in the human body is the superoxide anion. Not all reactive species are free radicals: for example, hydrogen peroxide (H2O2), which acts as a signalling molecule in several physiological pathways, can also be a precursor of free radicals, especially in the presence of transition metals (Fe++/Fe+++; Cu+).6
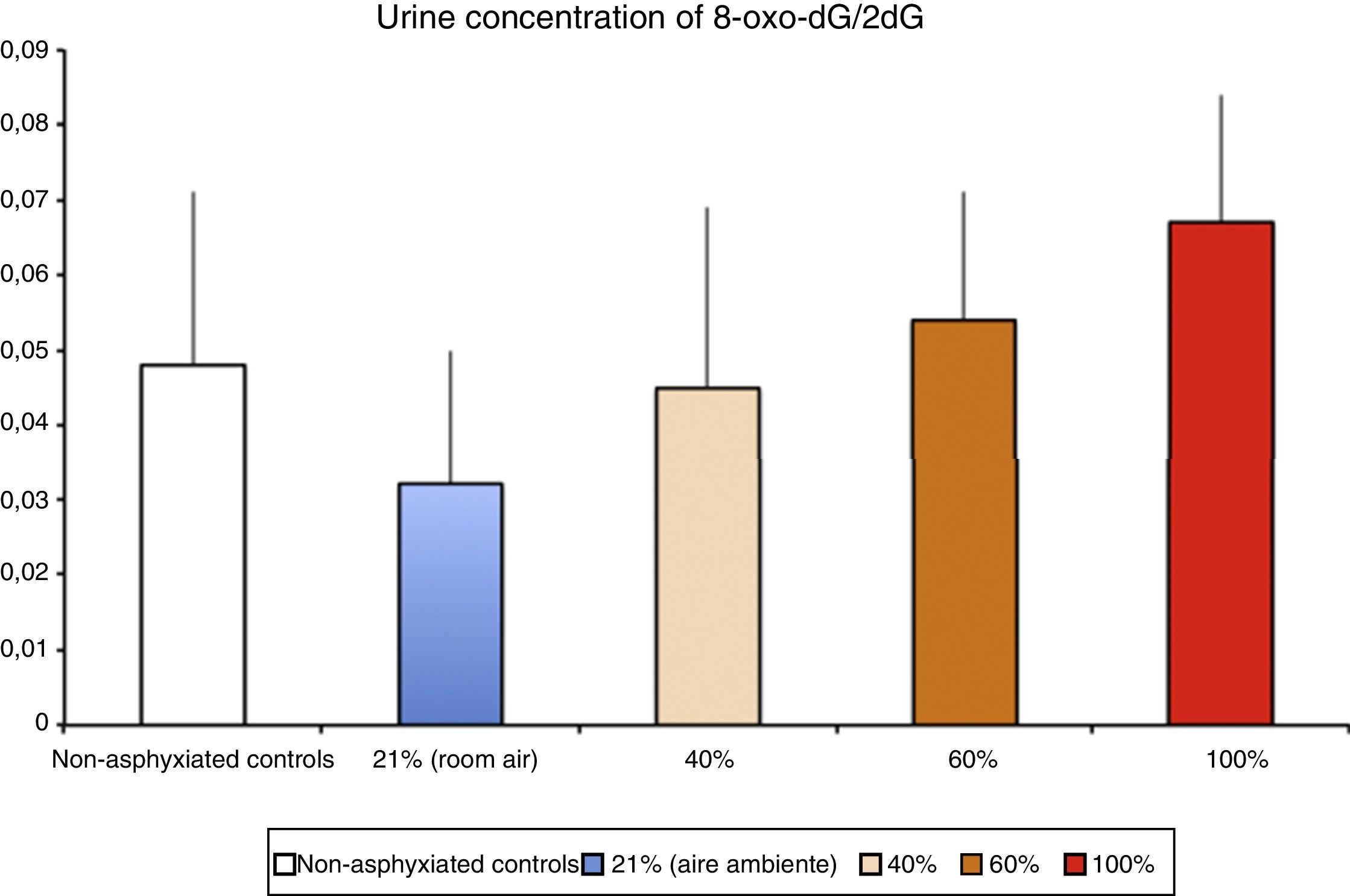
Levels of biomarkers associated with DNA damage in a newborn piglet model of hypoxia/reoxygenation correlated to the fraction of inspired oxygen used during reoxygenation (Solberg et al.17).
Mitochondrial activity is the main physiological source of free radicals. Within the mitochondria, oxygen is fully reduced and gives rise to water if it reacts with protons (H+). Under metabolic stress conditions (resuscitation, infection, parenteral nutrition, etc.) there is an increased production of free radicals that can damage mitochondrial structure and hinder energy production. Some of the most frequent causes of oxidative stress are the activation of the NADPH oxidase (NOX) system in phagocytes in response to infection, oxygen supplementation or the administration of certain drugs.6,8,9
Biological antioxidant systemsOxidative stress reflects a disturbance in the balance between the production of free radicals and their neutralisation. A certain degree of oxidative stress is required for proper functioning of enzymatic pathways that require reactive oxygen species for cellular signalling. However, a pronounced imbalance leads to tissue damage and acute or chronic disease.9
The antioxidant defence system (ADS) includes enzymatic as well as non-enzymatic systems. Antioxidant enzymes neutralise reactive species through chemical reactions. Non-enzymatic antioxidant systems include proteins that bind transition metals (transferrin, ceruloplasmin, ferritin), vitamins that block lipid peroxidation (A, E and C) and low molecular weight compounds that reduce reactive species. Reduced glutathione (GSH) reacts with another molecule of GSH to form oxidised glutathione (GSSG), a process that releases two electrons that can be captured by free radicals, contributing to their neutralisation. The enzyme GHS reductase then catalyses the reduction of GSSG to GSH. Some of the key antioxidant enzymes are superoxide dismutases (SODs), which catalyse the conversion of superoxide to hydrogen peroxide, and catalases (CATs) and glutathione peroxidases (GPxs), which convert hydrogen peroxide to water and molecular oxygen. Other systems involve glutaredoxins, thioredoxins, peroxiredoxins and haem oxygenases, which degrade the pro-oxidant haem complex in red blood cells, converting it to biliverdin. The ADS develops late in gestation to prepare the foetus for postnatal oxygenation.6,9
Oxidative stress, tissue damage, and their assessmentFree radicals cause changes in proteins, lipids, carbohydrates, RNA and DNA. There are biomarkers of overall redox status, such as the oxidised-reduced glutathione ratio (GSH/GSSG). In situations of oxidative stress, the level of GSSG increases and the ratio decreases, while the opposite occurs in situations promoting antioxidant activity or reduction, where there is an increase in GSH. Another method is the measurement of antioxidant enzymes. Thus, increases in oxidative stress are associated with a proportional increase in protective enzyme activity. Nevertheless, research is currently underway to search for biomarkers derived from the oxidation of different cell components that could be detected in samples of biological fluids. In recent years, the use of several biomarkers from different biological fluids for the detection of oxidative stress has been validated in newborns.10–16
Pathophysiology of ischaemia-reperfusion injuryFree radical generation in reoxygenation injuryHypoxia-ischaemia results in depletion of ATP, and this causes damage that is initially reversible but that cannot be repaired if the depletion is prolonged. Oxy-regulator tissues, of which the CNS is the prime example, are most sensitive to the lack of oxygen. Studies conducted in the 1970s proved that the initial damage caused by a period of hypoxia/ischaemia was amplified significantly during reperfusion/reoxygenation. The damage caused by reoxygenation was directly proportional to the duration and intensity of hypoxia-ischaemia and to the oxygen concentration used during reoxygenation/reperfusion.8 An experimental animal model of hypoxia-ischaemia in newborn piglets found that nervous tissue damage and the urine concentration of markers of DNA damage (Fig. 3) were proportional to the FiO2 used during reoxygenation.17 Research is currently being conducted on specific markers of tissue hypoxia. Lactate is a reliable marker of the intensity of tissue hypoxia, but does not reflect its duration. An experimental model in newborn piglets demonstrated that the concentrations of metabolites resulting from the oxidation of purines, pyrimidines and phospholipids detected by mass spectrometry can be used to calculate a “metabolite index” that provides a reliable and reproducible measure of the intensity and duration of brain hypoxia, which can be used for the purpose of prognosis or to guide decision-making regarding the most appropriate treatment.18–20
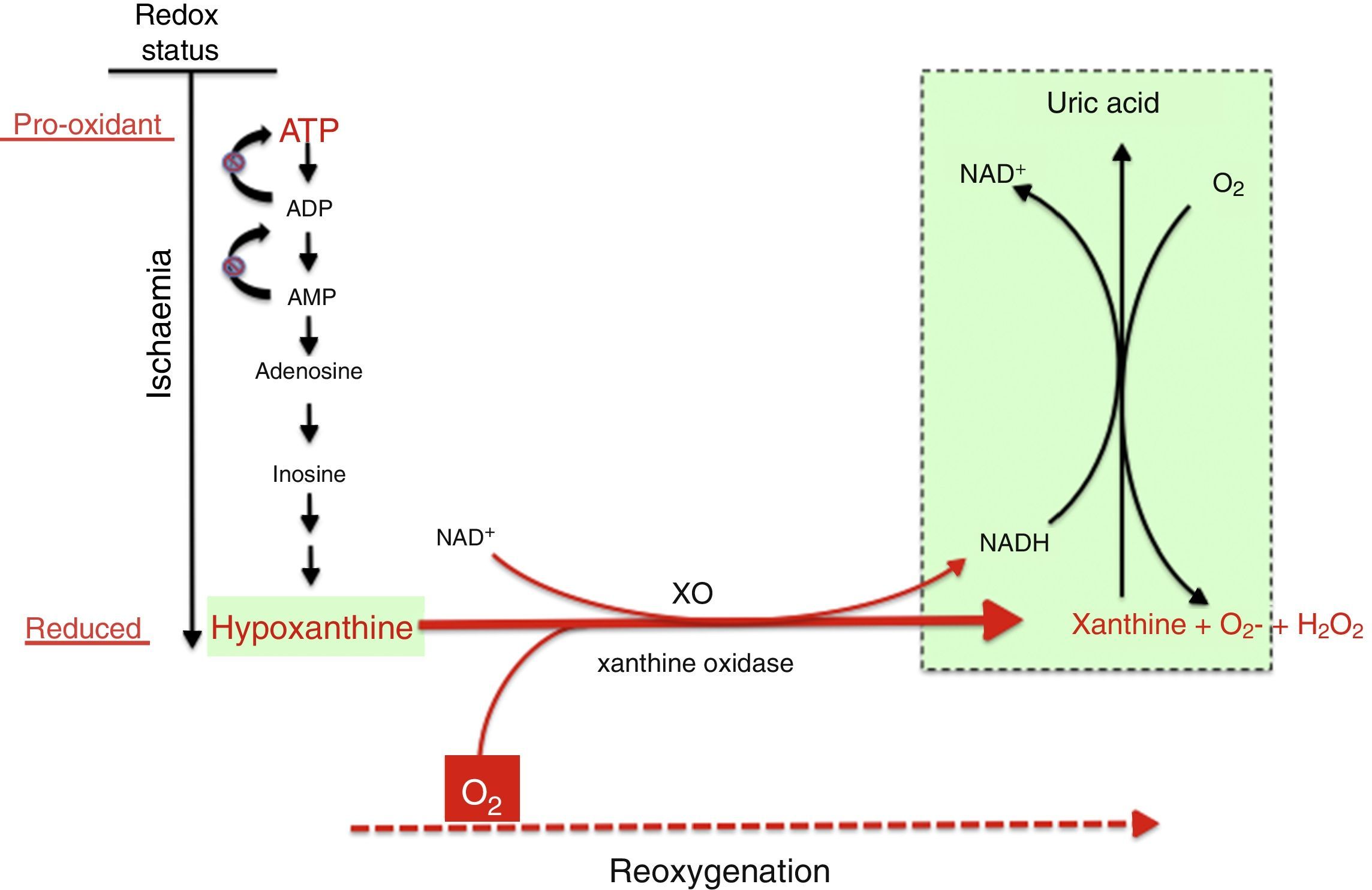
Mechanism of free radical generation by the action of the xanthine oxidoreductase complex. In prolonged hypoxia, there is an accumulation of purines from the degradation of ATP, especially hypoxanthine. During reoxygenation, xanthine reductase is converted to xanthine oxidase, which generates a large amount of reactive oxygen species, leading to oxidative stress.
There are multiple sources of reactive species during ischaemia-reperfusion (IR), chief of which are the mitochondrial respiratory chain complex and the xanthine oxidase (XO) system. The NADPH oxidase (NOX) system and NO synthase uncoupling are also involved, although their roles are less prominent.8
Under normal conditions, the production of free radicals is well regulated. Nearly all electrons that flow down the electron transport chain are captured by oxygen (98%), while only 2% produce reactive species. However, under hypoxic conditions, the transfer of electrons in the respiratory chain halts due to the lack of oxygen, and Ψm drops. Membrane depolarization inhibits the activity of ATP synthase and ATP production stops, leading to the lysis of mitochondria and cell death. During reoxygenation, the Ψm is re-established and the synthesis of ATP resumes. However, the abrupt reactivation of the electron flow leads to an excessive production of reactive species that may aggravate the neural tissue damage initially caused by apoptosis.2,7,8Fig. 4 shows electron microscopy images of brain slices from an experimental murine model of hypoxia-reperfusion that reveal the structural disruption, swelling, loss of cristae, membrane rupture and finally vacuolation and destruction of mitochondria.
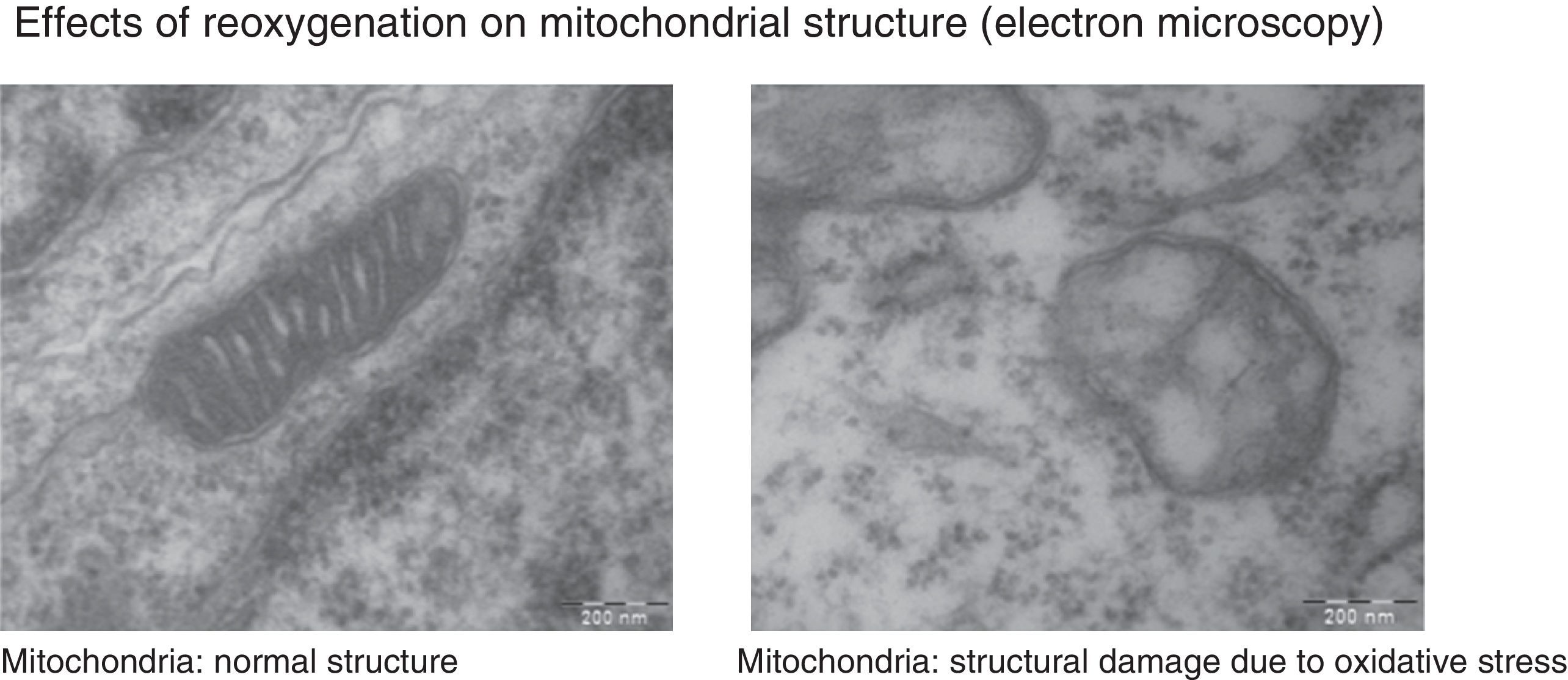
The images show morphological and structural changes including structural disruption, swelling, loss of cristae, membrane rupture and finally vacuolation and destruction of cerebral mitochondria in a model where newborn mice were exposed to different oxygen concentrations. Courtesy of Isabel Torres-Cuevas (doctoral thesis; supervisor: Máximo Vento).
In the reoxygenation stage of IR, oxygen reacts with hypoxanthine and XO, driving a burst of superoxide radicals and hydrogen peroxide that in turn give rise to other reactive species in a chain reaction, amplifying the tissue damage initially caused by prolonged ischaemia. The activity of XO is also subject to post-translational regulation by the partial pressure of oxygen in tissue, with XO activity increasing with decreasing pressure. This would explain how oxidative stress could begin in the hypoxaemic stage and be amplified during reoxygenation. Recent studies have demonstrated that XO can produce NO from nitrites. The simultaneous generation of superoxide and NO may result in the production of peroxynitrite, a powerful nitrating agent.21,22
Phases of hypoxic-ischaemic encephalopathyThe course of HIE can be divided into 3 phases (Fig. 5), although in reality it is a continuum in which these events can overlap. The first phase lasts 6h and is characterised by a reduction in blood flow in the foetus, causing systemic hypotension and loss of autoregulation of cerebral blood flow. Cerebral ischaemia leads to hypoxia, acidosis and brain damage as a result of primary energy failure. The cell resorts to anaerobic metabolism to compensate for the decreased oxygen supply, which causes lactic acidosis, ATP depletion, intracellular accumulation of Na+, Ca++ and water and inhibition of neurotransmitter reuptake with secondary excitotoxicity. The massive influx of Ca++ leads to activation of lipases and NO synthase, free radical production, mitochondrial dysfunction and release of apoptogenic substances to the cytoplasm. The second phase (6–48h) is characterised by continued excitotoxicity, loss of mitochondrial activity and oxidative stress secondary to changes in membrane potential with a reduced production of ATP and an alkaline intracellular environment despite adequate oxygenation. Finally, there is a third phase that may last days, weeks or even months characterised by inflammation and epigenetic changes that lead to abnormalities in axonal development, neurogenesis and synaptogenesis.23,24
Strategies to prevent damage from reactive oxygen species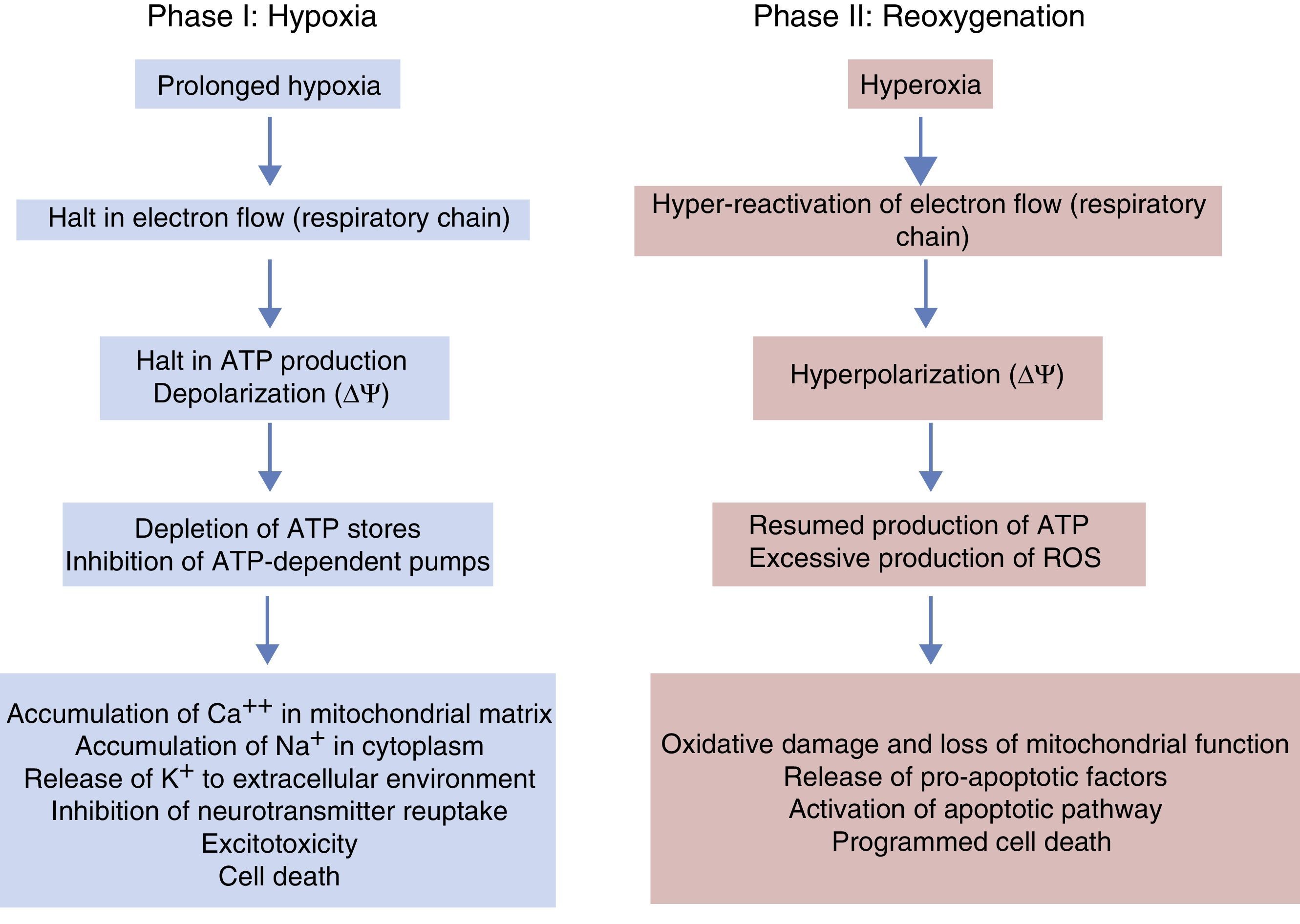
Preventing the damage caused by reactive oxygen species requires very early intervention, as the damage may have started in utero and could be amplified during postnatal resuscitation. Therefore, the treatment protocol should include interventions that target primary energy failure. These could be implemented in the mother during labour if there were evidence of foetal distress, or in newborns with clinical or electrophysiological findings suggestive of HIE in the first minutes of life.25,26
We proceed to give a brief description of interventions that address the pathogenic effects of oxidative stress.
HypothermiaHypothermia slows down the metabolic activity of tissues such as the CNS, with a corresponding reduction in ion exchange activity and ATP demand. The reduction in mitochondrial activity combined with activation of uncoupling proteins (UCPs) significantly decreases production of reactive species while preserving the membrane potential and preventing release of apoptogenic proteins. There is an extensive body of research on the mechanism of action of hypothermia and its clinical effectiveness. However, despite the proven efficacy of hypothermia as a neuroprotective intervention in patients with moderate or severe HIE, up to 45% of treated patients, especially those with severe HIE, have unfavourable outcomes. This may change in the very near future with the use of adjuvant therapies that are currently being investigated in experiments and clinical trials.5,23,25,27
Neonatal resuscitation with low oxygen concentrationsThe use of low oxygen concentrations during resuscitation is associated with a reduction in oxidative stress during reoxygenation, mortality, and the incidence of HIE.28 The recent guidelines of the International Liaison Committee on Resuscitation published in 2015 recommend the use of room air in the resuscitation of term newborns.29
Allopurinol or inhibition of xanthine oxidaseAllopurinol is a selective inhibitor of XO, reduces production of NO from nitrites and acts as a free-iron chelator and a hydroxyl radical scavenger. These properties make it a suitable candidate for neuroprotection.21,22
A pilot study was conducted in 199830 with administration of 40mg/kg of allopurinol in asphyxiated newborns. The study found a reduction in oxidative stress and improved cerebral perfusion and cortical activity in the absence of allopurinol-related toxicity. However, a subsequent study31 found no improvements in survival or neurodevelopmental outcome with administration of allopurinol in the first 4h post birth compared to the use of placebo, which the authors attributed to the delay in its administration after birth. The same group of researchers administered allopurinol to pregnant women in labour with signs of foetal hypoxia.32 Newborns who had therapeutic concentrations of allopurinol in cord blood had significantly lower plasma levels of S-100B protein, a marker of brain damage, and non-protein-bound iron, a marker of oxidative stress.32 A follow-up study of moderately asphyxiated newborns found that the group treated with allopurinol in the first hours post birth (<4h) and a second dose at 12h scored higher in the Wechsler Intelligence Scale for Children (WISC) compared to newborns that received placebo.33 Recently, the same authors published the results of a randomised placebo-controlled multicentre trial of allopurinol in women in labour with clinical indices of foetal hypoxia prompting immediate delivery, which did not confirm a significant reduction in the levels of S100B; however, the post hoc analysis revealed a potential benefit in newborns of mothers treated with allopurinol.34
A multicentre study is currently being conducted in Europe (ALBINO trial; EudraCT-2016-000222-19) with participation of 18 public hospitals in Spain. It is a phase III randomised placebo-controlled trial of allopurinol whose primary endpoint is death and/or severe neurodevelopmental impairment. The protocol consists of administration of a dose of allopurinol to newborns with suspected HIE secondary to neonatal asphyxia in the first 30minutes of life, with a second dose administered 12h after the first in newborns treated with hypothermia. The assessment includes metabolomic, neurophysiological and imaging tests, in addition to clinical followup of participants until age 24 months.
MelatoninMelatonin (N-acetyl-5-methoxytryptamine) is an endogenous indolamine with antioxidant, anti-inflammatory and antiapoptotic properties that has shown promising results in patients with HIE. Its anti-inflammatory activity is based on the downregulation of pro-inflammatory molecules and the suppression of NO production in neural tissue. Melatonin also blocks the release of pro-apoptotic proteins by mitochondria and modulates the activity of GABA and glutamate receptors.35 Melatonin used as adjuvant therapy to hypothermia in patients with HIE is associated with a reduction in oxidative stress and improvements in survival and neurodevelopmental outcomes,36–38 although these results are limited by the small sample sizes. Further studies with adequate statistical power are required to confirm the effectiveness of melatonin and address additional questions.
ConclusionsHypoxic-ischaemic encephalopathy continues to be a serious challenge in neonatology. Despite the widespread use of hypothermia, the associated morbidity and mortality continue to be high. Reactive oxygen species play a significant role in the pathogenesis of asphyxia leading to HIE. Therefore, a reduction of their production, their neutralisation or the inhibition of the inflammatory and pro-apoptotic pathways that they activate are strategies that combined with interventions involving other targets (for instance, to reduce excitotoxicity or promote angiogenesis or neurogenesis) can improve intact survival in newborns with HIE.
FundingThis study has been funded by Grant EC11-244 (Instituto de Salud Carlos III; Ministry of Economy, Industry and Competitiveness; Spain) awarded to MV and through a contract between AN and the Maternal and Child Health and Development Network (SAMID), funded by the 2008–2011 R&D&I National Programme of the General Vice Directorate of the Instituto de Salud Carlos III and project RD16/0022/0001 of the European Regional Development Fund.
Conflicts of interestThe authors have no conflicts of interest to declare.
List of hospitals and co-researchers that participated in the EC11-244 clinical trial (Instituto de Salud Carlos III; Ministry of Economy, Industry and Competitiveness; Spain), with registration number EudraCT-2011-005696-17
Hospital Universitario y Politécnico La Fe, Valencia: Antonio Nuñez-Ramiro; Anna Parra-Llorca; Ana García-Robles; Ana García-Blanco; María Cernada; Raquel Escrig-Fernández; Ana Gimeno-Navarro; Nuria Boronat González.
Hospital Universitario Puerta del Mar, Cadiz: Isabel Benavente; Simón Lubián López.
Hospital Regional Universitario Materno Infantil Carlos Haya, Malaga: Mercedes Chaffanel; Enrique Salguero; María del Mar Serrano Martín; Rafael Maese Heredia.
Hospital Universitario Gregorio Marañón, Madrid: Dorotea Blanco; María Arriaga.
Hospital Universitario La Paz, Madrid: Eva Valverde; Malaika Cordeiro; Adelina Pellicer; Laura Sánchez; Paloma López Ortego; Maricarmen Bravo.
Hospital Universitario 12 de Octubre, Madrid: María Teresa Moral-Pumarega; María del Carmen Pérez-Grande; Noelia Ureta Velasco; Rocío Mosqueda Peña; Ana Melgar Bonis.
Hospital Universitario de Cruces, Barakaldo: Begoña Loureiro; Jon López de Heredia; María Jesús Martinez; Cristina Iniesta.
Hospital Universitario Vall d’Hebron, Barcelona: Héctor Boix; Yolanda Castilla; Cristina Fernández-García.
Complejo Hospitalario Universitario Vigo, Vigo: Juan Fernández-Lorenzo; Eva González-Colmenero; María Luisa González-Durán; Ana Concheiro-Guisán.
Hospital Universitario Central de Asturias: Belén Fernández Colomer; Rosa Patricia Arias Llorente.
Hospital Universitario Virgen del Rocío, Seville: Antonio Pavón; José Fernando Ferreira López.
Hospital Universitario Reina Sofía, Cordoba: Inés Tofé; Pilar Jaraba.
Hospital Universitario Quironsalud, Madrid: Fernando Cabañas; M. Angeles Caballero; Manuela López-Azorín.
Please cite this article as: Nuñez A, Benavente I, Blanco D, Boix H, Cabañas F, Chaffanel M, et al. Estrés oxidativo en la asfixia perinatal y la encefalopatía hipóxico-isquémica. An Pediatr (Barc). 2018;88:228.e1–228.e9.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals