


We present the case of a female full-term neonate with an adequate birth weight. The mother was 28 years old, secundigravida (previous spontaneous miscarriage in the first trimester) and had a history of type 2 diabetes mellitus treated with insulin and rheumatoid arthritis in treatment with a biologic (certolizumab). The maternal grandparents were from Pakistan and consanguineous (first cousins). Two maternal first cousins had died before age 2 years, with no other relevant family history.
The prenatal ultrasound scans revealed intrauterine growth restriction with normal blood flow parameters on Doppler and no other abnormal findings.
The birth was uncomplicated and the patient had Apgar scores of 9/10/10 and a normal cord blood pH (venous pH, 7.31). At 18 hours post birth she became hypoactive, with expiratory grunt, hypoglycaemia (blood glucose, 35 mg/dL) and sustained hypoxaemia (basal saturations of 90%–92%), prompting admission to the neonatal unit.
The initial blood panel revealed severe metabolic acidosis with hyperlacticaemia (peaking at 22 mmol/L at 21 h post birth). Infection was ruled out and, due to the suspicion of mitochondrial disease, treatment initiated with cofactors via the oral route: thiamine, biotin, riboflavin, coenzyme Q10 and L-carnitine.
The amino acid and organic acid profile revealed lactic acidaemia with elevation of alanine and proline.
The cardiological evaluation detected severe left ventricular systolic dysfunction with an ejection fraction of 37%. The patient received support with milrinone and furosemide and responded well, with resolution of metabolic acidosis and a decrease in lactate levels to less than 5 mmol/L. The dose of milrinone could be tapered off and eventually discontinued after initiation of carvedilol. Later on, captopril was added and maintained until hospital discharge.
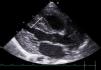
The standardised echocardiographic assessments evinced normalisation of systolic function in both ventricles and structural improvement, albeit with persistent thickening of the left ventricle and trabeculation in the right ventricle (Fig. 1).

The extended evaluation also detected bilateral blue-dot cataracts.
The presence of the clinical triad of lactic acidosis, cataracts and cardiomyopathy (probably progressing to hypertrophic cardiomyopathy given the thickening of left ventricle) led to suspicion of Sengers syndrome, which was confirmed through clinical exome sequencing. The analysis detected a homozygous pathogenic nonsense variant in the AGK gene (NM_018238.3: c.632G>A).
The patient maintained adequate weight gain and linear growth with mixed feeding and, given the favourable course, was discharged from the neonatal unit at 38 days post birth under treatment with carvedilol, captopril and furosemide. At the time of hospital discharge, she required a nasogastric tube to complete feedings in order to avoid muscle fatigue that could give rise to hyperlactataemia.
The patient remained in multidisciplinary follow-up with the support of the chronic complex disease unit.
During the follow-up by paediatric cardiology, there was evidence of progression to hypertrophic cardiomyopathy, which was managed medically with the treatment described above.
The patient showed signs of limited visual acuity secondary to the bilateral congenital cataracts. Given her haemodynamic stability, which suggested a longer life expectancy than initially expected, and in order to improve her quality of life and her interactions with the environment, the decision was made by a multidisciplinary team to perform cataract surgery at age 6 months. The patient died from cardiac arrest in the postoperative period (more than 24 hours after the procedure).
Sengers syndrome is a rare disease, with approximately 40 cases described to date. It is an autosomal recessive mitochondrial disease caused by biallelic pathogenic variants in the AGK gene encoding the mitochondrial enzyme acylglycerol kinase. The protein it encodes is part of the mitochondrial membrane and the TMEM22 complex. It is involved in signalling, mitochondrial DNA stability and mitochondrial energy metabolism through the mitochondrial respiratory chain. It plays a crucial role in the synthesis of lipids such as cardiolipin. In fact, a drug that stabilises mitochondrial structure via cardiolipin has been studied as a potential treatment option for this defect (elamipretide).1–3
The syndrome is characterized by lactic acidosis, hypertrophic cardiomyopathy and bilateral cataracts, with or without skeletal myopathy. There are 2 forms of the disease: (1) a severe infantile form characterised by early-onset cardiomyopathy, lactic acidosis and death in infancy, as observed in our patient, and (2) a milder form with a life expectancy that in some cases reaches into the forties in which cardiomyopathy develops at later stages.1,2
Although a clear genotype-phenotype correlation has yet to be established, there are reports that the presence of homozygous pathogenic nonsense variants, as was the case of our patient, is associated with a phenotype characterised by a high mortality in infancy and a shorter life expectancy.
The use of anaesthesia carries a high risk of sudden death due to the inhibition of mitochondrial metabolism by anaesthetic agents and the underlying cardiac dysfunction.4,5
First-year mortality reaches 50%, chiefly on account of heart failure. It is important for clinicians to suspect this syndrome in the presence of the described triad given the prognostic implications and for the purpose of genetic counselling.
Previous meeting: Presented at the 26th Annual Meeting of the Sociedad Catalana de Pediatría as a brief oral communication, Hiperlactacidemia en el neonato, ¿en qué debemos pensar?; June 10–11, 2022; Lérida, Catalonia, Spain.
