



Microvillous inclusion disease is a rare autosomal recessive condition, characterized by severe secretory diarrhoea that produces a permanent intestinal failure and dependency on parenteral nutrition. It usually begins in the neonatal period, and the only treatment at present is intestinal transplantation.
Patients and methodsA retrospective review was conducted on 6 patients (three males and three females) diagnosed with microvillous inclusion disease between 1998 and 2013.
ResultsAll debuted in the first month of life, with a median age of three days (range, 3–30 days), and had secretory diarrhoea dependent on parenteral nutrition, with fasting faecal volume of 150–200ml/kg/day. Light microscopy of duodenal biopsy samples showed varying degrees of villous atrophy without cryptic hyperplasia, accumulation of PAS positive material in the cytoplasm of enterocytes brush border, and anti-CD10 immunostaining was suggestive of intracytoplasmic inclusions. Diagnostic confirmation was performed with electron microscopy. Two of them had a genetic study, and showed mutations in MYO5B gene. Three died and three are alive; two of them with an intestinal transplantation and the third waiting for a multivisceral transplantation.
La enfermedad por inclusiones microvellositarias es una entidad rara, de herencia autosómica recesiva y caracterizada por una diarrea grave de carácter secretor que produce un fracaso intestinal permanente dependiente de nutrición parenteral. Habitualmente se inicia en el período neonatal y el único tratamiento posible en el momento actual es el trasplante intestinal.
Pacientes y métodosSe revisa, de forma retrospectiva, a 6 pacientes (3 varones y 3 mujeres), diagnosticados entre 1998 y 2013 de enfermedad por inclusiones microvellositarias.
ResultadosTodos comenzaron en el primer mes de vida, con una mediana de edad de tres días (rango: 3-30 días) y presentaron diarrea secretora dependiente de nutrición parenteral, con un volumen faecal en ayunas de 150-200ml/kg/día. La microscopia óptica de muestras biópsicas duodenales mostró grados variables de atrofia vellositaria sin hiperplasia críptica, con acumulación de material PAS positivo en el citoplasma de los enterocitos del borde en cepillo y la inmunotinción anti-CD10 fue indicativa de inclusiones intracitoplasmáticas. La confirmación diagnóstica se realizó con microscopia electrónica. En 2 de ellos se realizó estudio genético que demostró mutaciones en el gen MYO5B. Evolutivamente, 3 fallecieron y 3 se encuentran vivos; 2 de ellos portadores de trasplante intestinal y el tercero en espera de trasplante multivisceral.
Microvillous inclusion disease (MVID) or congenital/familial microvillous atrophy is due to a congenital disorder of the epithelial cells of the bowel. The literature has reported multiple mutations in the MYO5B gene that encodes myosin Vb that are associated with the development of this disease. It usually manifests in the neonatal period with severe diarrhoea that persists despite fasting, with children becoming permanently dependent on parenteral nutrition (PN). Its histological features consist in the absence or altered appearance of the enterocyte brush border membrane along with the presence of characteristic microvillous inclusions. The treatment of MVID includes PN and intestinal transplantation.
Patients and methodsWe performed a retrospective review of the patients diagnosed with MVID in our hospital in a 15-year period (1998–2013). We analyzed epidemiological, clinical, diagnostic, treatment and patient outcome variables. Biopsy samples were collected by means of upper gastrointestinal endoscopy for diagnostic confirmation. A conventional histological examination was performed that included a haematoxylin–eosin stain (HE) and a periodic acid-Schiff stain (PAS), as well as immunohistochemical testing with anti-CD10 antibodies and electron microscopy examination. Two of the six patients underwent genetic testing for mutations in the MYO5B gene. Infectious, allergic and autoimmune aetiologies, defects in digestion, absorption and transport of nutrients and other diseases of the intestinal mucosa were ruled out in all patients.
ResultsThe onset of diarrhoea occurred within four days from birth in five patients, at a median age of 3 days (range, 3–30 days) (Table 1). Three patients were male and 3 were female. Patients 2 and 3 were siblings born to consanguineous parents, and there was no blood relationship between the two parents of the remaining patients. Patient 6 was born to term, while the rest were born at 35–36 weeks of gestational age. All were born with normal weights for their gestational age. Prenatal ultrasonography had shown dilation of bowel loops in half of the patients, with no other significant findings.
General characteristics of patients with microvillous inclusion disease.
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| Sex | M | M | F | F | F | M |
| Prenatal ultrasound | Normal | Normal | Normal | Bowel dilation | Bowel dilation | Bowel dilation |
| Birth weight (g) | 2980 | 2590 | 2660 | 2555 | 2320 | 3680 |
| GA (weeks) | 36 | 36 | 35 | 36 | 35 | 39 |
| Consanguinity | No | Yes | Yes | No | No | No |
| Age at onset (days) | 30 | 3 | 4 | 3 | 3 | 4 |
| Stool volume (ml/kg) | 90–120 | 200–300 | 120 | 150–200 | 150–200 | 200 |
| Faecal Na (mEq/L) | 112.8 | 81.18 | 116.3 | 106.16 | 113.16 | 104.18 |
| Faecal Na/K | 9.2 | 4.5 | 7.96 | 6.6 | 7 | 5.7 |
| Extraintestinal manifestations | No | No | No | No | Nephrocalcinosis, renal lithiasis, haematuria | Cholestasis, glomerulonephritis |
| MYO5B Mutation | NP | NP | NP | NP | Yes | Yes |
| Outcome | AliveITx15 age | Died of sepsis (4 months) | AliveLITx11 years | Died while on Tx list (16 months) | Died while on Tx list (29 months) | AliveIn multivisceral Tx list |
F, female; GA, gestational age; ITx: intestinal transplantation; LITx, liver and intestinal transplantation; M, male; NP, not performed.
All six patients had onset with severe diarrhoea (stool volume ∼120–200ml/kg/day) of the secretory type (sodium in stools >100mEq/L) and with abundant mucus, which persisted despite fasting. Patient 5 developed renal complications associated with PN and severe electrolyte imbalances, and patient 6 developed intense skin itching as a prominent symptom. None of the patients could tolerate any type of enteral formula, and all required PN to prevent dehydration and metabolic acidosis.
The upper gastrointestinal endoscopy did not show significant macroscopic abnormalities. The diagnosis was made by the histological examination of duodenal biopsies with light and electron microscopy. The presence of mutations in the MYO5B gene was confirmed in patients 5 and 6. The rest of the patients did not undergo genetic testing. All patients received PN in the hospital and at home for prolonged periods of time (3–36 months). Three of the six patients have died (one at age 3 months of septic shock, and the other two of liver complications associated with intestinal failure while they were on the transplant list). Of the three survivors, one is awaiting a multivisceral transplantation and has developed acute postinfectious glomerulonephritis, and the other two have undergone transplantation (one an intestinal transplantation and the other a combined liver and intestinal transplantation, 15 and 11 years ago, respectively), and have achieved intestinal autonomy and a good quality of life.
DiscussionThere are different causes of congenital diarrhoea with onset in the early months of life: defects in the digestion, absorption and transport of nutrients and electrolytes, defects of enterocyte differentiation and polarization, defects of enteroendocrine cells differentiation and defects in the modulation of the intestinal immune response.1 Chief among the congenital enterocyte disorders are intestinal epithelial dysplasia and MVID, which are rare causes of permanent intestinal failure.
Microvillus inclusion disease, also known as congenital microvillus atrophy, was first described by Davidson et al. in five patients in 1978,2 and the term MVID was proposed by Cutz et al. in 1989.3 It is a congenital disease that has been recently associated in the literature with mutations in the MYO5B gene of chromosome 18 that result in the altered expression of myosin Vb in enterocytes.4–7 Myosin Vb is a protein that binds with Rab proteins to provide the molecular basis for the regulation of recycling endosome pathways and their transport along actin filaments to the apical membrane in polarized epithelial cells. Homozygous as well as heterozygous mutations have been characterized that can cause MVID.4–7 The exact genetic alteration was not known until 2008.4,5 Different mutations in the MYO5B gene have since been described, with up to 41 reported until the present date.8 The two most recent patients in our series underwent genetic analysis of the MYO5B gene, and two heterozygous mutations were detected in each: the mutations c.3190C>T (p.R1064X) and c.3514C>T (p.Q1172X) in patient 5, and c.445C>T (p.Q149X) and c.5383C>T (p.R1795X) in patient 6. The patients that had been diagnosed before did not undergo genetic testing.
There are patients with MVID in whom testing has not revealed mutations in the MYO5B gene. Very recently, whole-exome sequencing of DNA from patients with MVID has shown homozygous mutations in syntaxin 3 in some.9 Syntaxin 3 is a receptor involved in the membrane fusion of apical vesicles in enterocytes, and mutations result in their defective fusion with the apical membrane. It has been proposed that this is an atypical MVID variant, with a milder phenotype, a delayed onset, greater tolerance of enteral nutrition and subtle histological differences.10 Still, more studies are needed to properly establish a correlation between different mutations and phenotypical expression.
During gestation, the presence of polyhydramnios and dilation of intestinal loops can be detected by ultrasonography, albeit irregularly.11 Three of our patients showed intestinal dilation without polyhydramnios in the third-trimester ultrasound. All prenatal ultrasound scans were normal in the other patients.
The disease manifests with intractable diarrhoea that usually starts in the first week of life (early-onset form) although cases with onset after weeks 6–8 of life have also been described (late-onset form).12–14 The diarrhoea is watery and abundant, with plenty of mucus, and can sometimes be mistaken with urine, with stool volumes of 100–200ml/kg/day even when the patient is fasting. It leads to significant dehydration in a few hours, requiring permanent PN and high volumes of fluids and electrolytes. All our patients presented with significant weight loss and metabolic acidosis, as well as a considerable loss of sodium and chloride in stools (around 100mmol/L and 80mmol/L, respectively). Patient 5 was transferred from another hospital with a tentative diagnosis of enterovesical fistula based on the stool characteristics.
Historically, it was believed that MVID was not usually associated with other extraintestinal malformations, but some authors have described a co-occurrence in some of their patients of heart malformations, Down syndrome, neuronal dysplasia, hypochondroplasia or sensory deficits among others.14–19 Despite the widespread expression of the MYO5B gene in all epithelial tissues, it is not clear how organs other than the intestine may be affected by this disease. There has been evidence in recent years that extraintestinal manifestations are not as rare as initially thought. A recently published series of 24 patients with MVID followed up at ages ranging from birth to age 23.5 years (median duration of followup, 4.7 years) found liver disease in 22 patients, kidney disease in nine, and pulmonary disease in two.19
Liver diseases, such as cholestasis, cholelithiasis and liver fibrosis, may result from prolonged PN. However, slightly elevated or normal levels of gamma-glutamyl transpeptidase (GGT) indicate that NP cannot be the sole cause of liver dysfunction. A specific form of liver disease associated to MVID has been described that is characterized by normal GGT levels and is similar to progressive familial intrahepatic cholestasis (PFIC).19 Myosin Vb is needed for bile canalicular formation and also for the trafficking of ATP-binding-cassette transporters.20 In a study of a cohort of 28 patients with MVID of whom 8 developed cholestasis either pre or post transplant and with clinical and laboratory findings similar to those of PFIC, Girard et al. concluded that cholestasis in these patients could result from impairment of the MYO5B/RAB11A apical recycling endosome pathway in hepatocytes, altered targeting of bile salt export pumps (BSEPs) to the canalicular membrane, and increased ileal bile acid absorption.21 It is not known why not all patients develop cholestasis. Patient 6 in our series had significant cholestasis for 9 months that manifested with intense cutaneous itching and normal GGT levels. A liver biopsy of the patient did not show significant histological abnormalities, and immunohistochemical testing ruled out a BSEP and a MDR3 deficiency. The cholestasis has since resolved and the patient is now a candidate for multivisceral transplantation. It has been proposed that patients that have had liver involvement pre transplantation undergo a combined liver and intestinal transplantation to avoid the development of cholestatic diseases in the future.
An association with kidney symptoms, such as haematuria or Fanconi syndrome, has also been described, 22 as well as with endocrine conditions like diabetes.23 Patient 5 developed nephrocalcinosis, lithiasis and haematuria, and patient 6 acute postinfectious hypocomplementemic glomerulonephritis.
The diagnosis of MVID is made by finding the histological features characteristic of the disease in a duodenal or jejunal biopsy specimen. While light microscopy can guide the diagnosis, the definitive diagnosis requires electron microscopy.24,25 The histological abnormalities may be found in the colon13 as well as the gastric mucosa.26 The presence of mutations in the MYO5B gene completes the diagnosis.
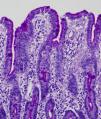
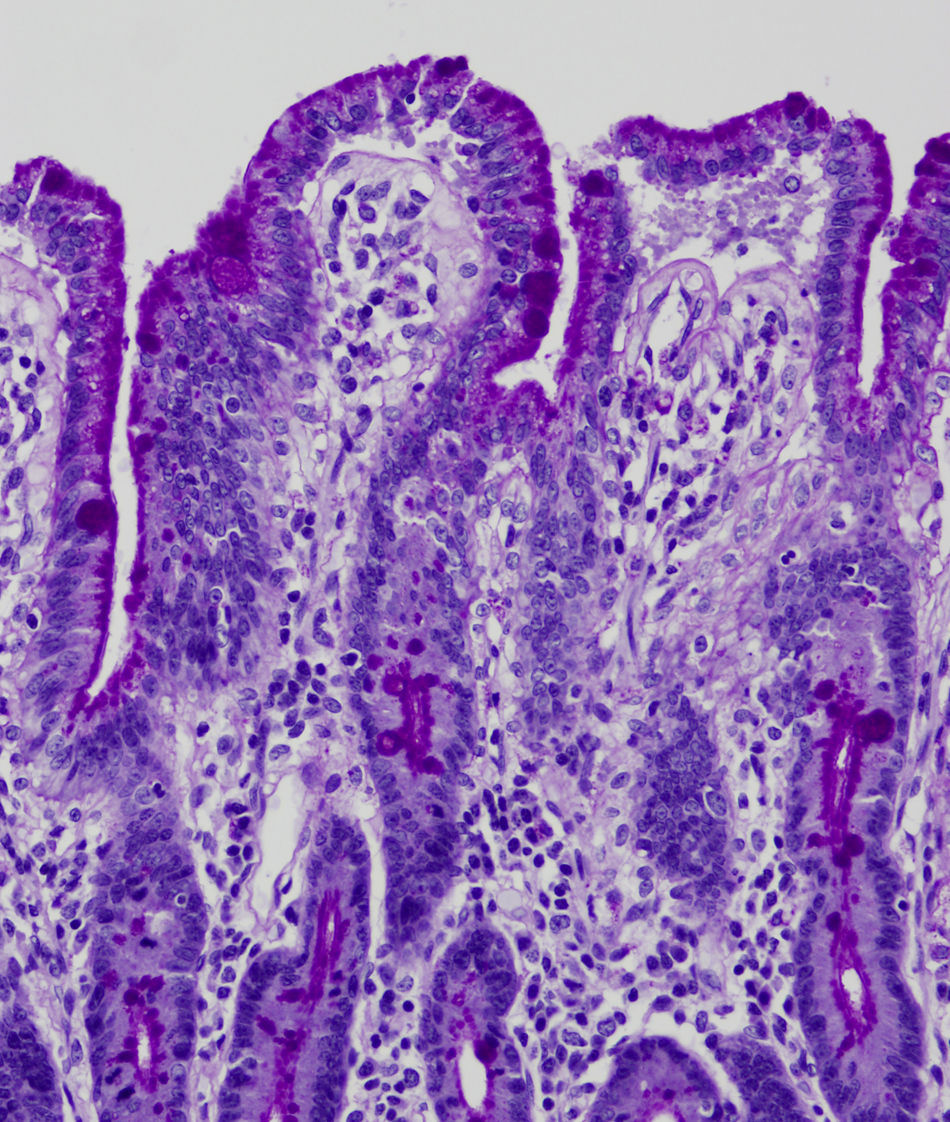
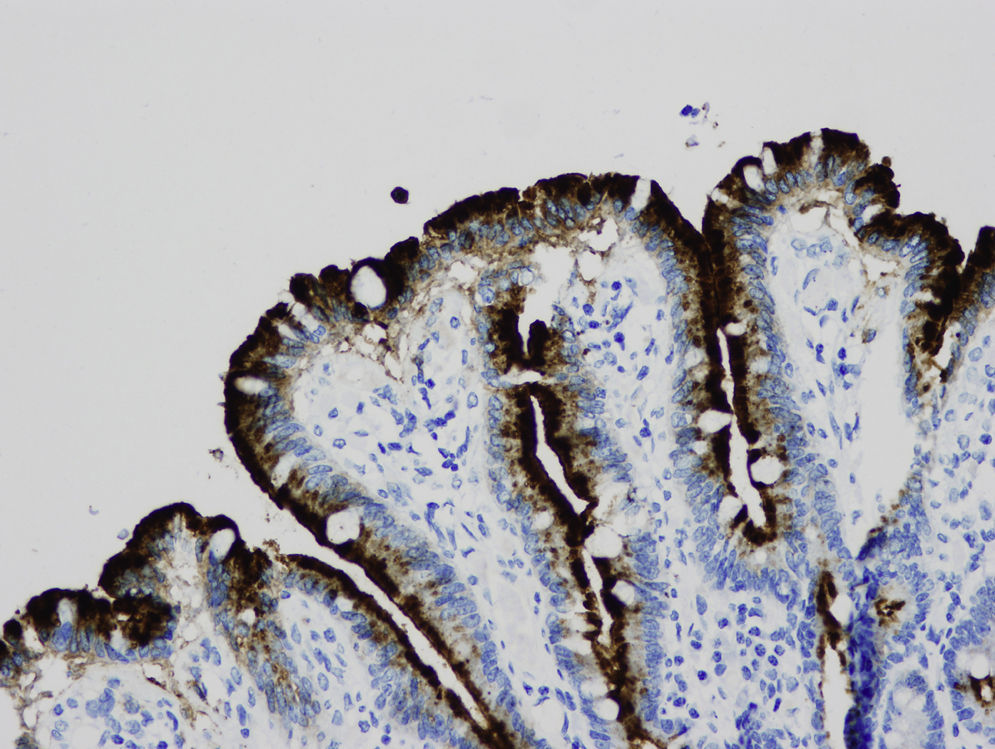
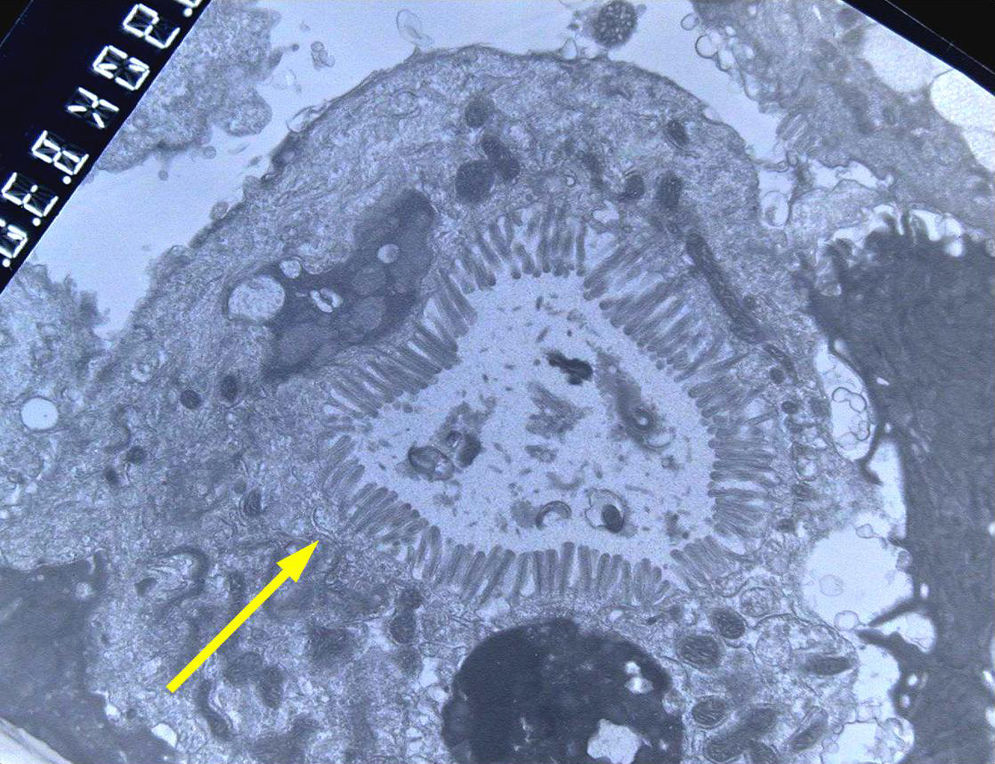
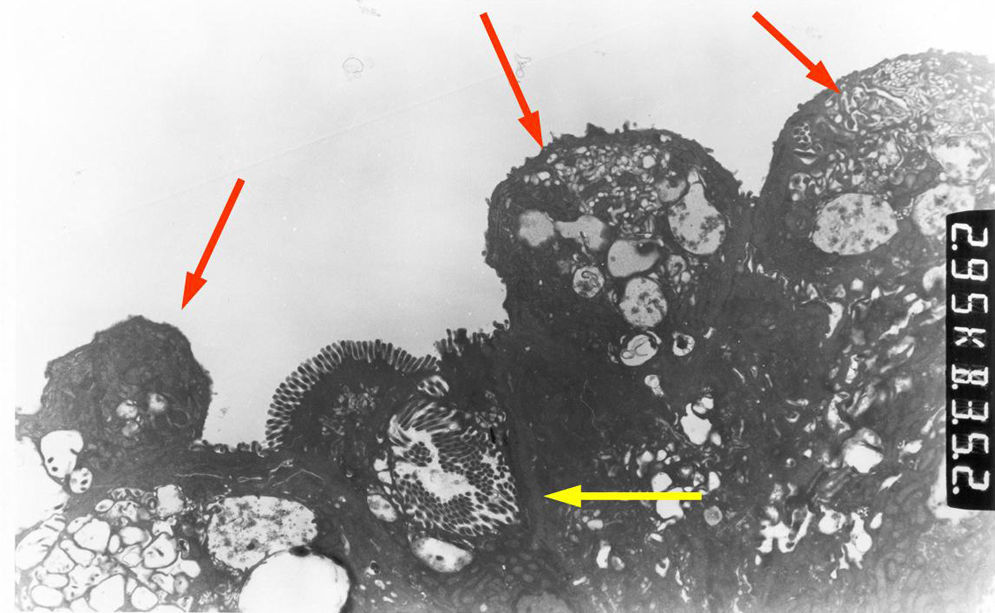
Light microscopy shows varying degrees of atrophy of the brush border microvilli, no significant signs of an inflammatory response and a normal or slightly increased density of the crypts. In healthy subjects, alkaline phosphatase27 and PAS staining only stain the brush border of the enterocyte, while in patients with MVID the border is either not stained or is stained unevenly and there is an accumulation of PAS-positive materials in the apical cytoplasm of enterocytes (Fig. 1). This can also be observed by means of CD1028 (Fig. 2) and carcinoembryonic antigen immunostaining.29 The three key ultrastructural findings include the reduction, absence and/or disorganization of microvilli in mature enterocytes; the accumulation in the apical cytoplasm, and especially of crypt cells, of secretory granules; and the characteristic intracytoplasmic inclusion bodies lined with microvilli (Fig. 3). However, it is not always easy to make a histopathological diagnosis of MVID due to the existence of variants, different clinical manifestations and even a small percentage of patients in whom the typical inclusion bodies cannot be detected. In 1994 Raafat et al.30 described three children with microvillous dystrophy, a condition they considered a clinicopathologic variant of MVID that has since been addressed by other authors.24 This ultrastructural lesion was observed in patient 1 (Fig. 4).
Disease progression invariably results in the need for PN, since enteral nutrition is not possible due to the large intestinal losses that carry the risk of dehydration and death.
Pharmacological treatment has been attempted with various agents, such as loperamide, somatostatin, corticosteroids or growth factors, among others, but none of them were efficacious in these patients.30,31 Intestinal transplantation is currently the only option available to patients with permanent intestinal failure, especially those experiencing difficulties in the administration of PN (loss of venous access due to deep vein thrombosis) or severe complications from PN (liver disease, sepsis secondary to the presence of a central line, etc.).19,32,33 Two of our patients received transplants and had favourable outcomes; they have achieved intestinal autonomy and have not experienced complications associated with their underlying disease. A third patient is waiting for a multivisceral transplantation, although at present he has been temporarily removed from the transplant list due to glomerulonephritis.
There have been isolated cases in the literature in which the disease improved and the patient achieved intestinal autonomy and could discontinue PN.16,34 In a recent review of 8 cases of MVID with atypical manifestations (delayed onset and smaller diarrhoea volumes), Perry et al.14 described 3 cases with favourable outcomes that allowed the discontinuation of PN. In light of this, they proposed expanding the clinical spectrum of this disease to include patients with milder forms, which entails that MVID should be considered in the differential diagnosis of intractable diarrhoea beyond the neonatal period.
The most common causes of death are severe metabolic decompensation or complications from PN (liver disease or sepsis), as occurred in the three patients that died in our series.
In brief, MVID is a severe inherited genetic disease that causes early intestinal failure dependent on PN and can only be cured with an intestinal transplant. It is diagnosed on the basis of clinical, histological, ultrastructural and genetic findings, but diagnosis may be complicated in cases with atypical presentations or with an incomplete clinical picture. A differential diagnosis must be made to rule out other causes of early-onset severe diarrhoea.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Fernández Caamaño B, Quiles Blanco MJ, Fernández Tomé L, Burgos Lizáldez E, Sarría Osés J, Molina Arias M, et al. Fracaso intestinal y trasplante en la enfermedadpor inclusiones microvellositarias. An Pediatr (Barc). 2015;83:160–165.
