





To determine the incidence of spinal muscular atrophy (SMA) in our study population and genetic distribution and epidemiological and clinical characteristics and to analyse the level of care and development.
Material and methodRetrospective descriptive study of patients treated in our hospital in the past 25 years (from 1987 to early 2013), with a clinical and neurophysiological diagnosis of SMA.
ResultsA total of 37 patients were found, representing an incidence for our reference population and year of 1 case per 10,000 live births. Males predominated (male/female ratio: 1.6/1). The type of SMA diagnosed more frequently was type I (26 cases), followed by type II (9 cases), one case with SMA type III, and one case of spinal muscular atrophy with respiratory distress type 1 (SMARD1). The most frequent genetic alteration was homozygous deletion of exons 7 and 8 of SMN1 gene in 31 cases, while five patients had atypical genetics. The median survival for type I was 8.0 months and 15.8 years for type ii.
ConclusionsThe incidence in our population remains stable at around 1/10,000. Most cases presented with, predominantly male, typical genetics. In approximately 1/10 patients the genetic alteration was different from the classical one to the SMN gene. The prevalence of AME unrelated SMN gene was 1/37. The level of care has increased in line with social and welfare demands in recent years.
Conocer la incidencia de la atrofia espinal infantil (AME) en nuestra población y estudiar la distribución genética y las características epidemiológicas y clínicas, el nivel de cuidados y su evolución.
Material y métodoEstudio descriptivo retrospectivo de los pacientes atendidos en nuestro hospital en los últimos 25 años (1987–2013), con diagnóstico clínico y neurofisiológico de AME.
ResultadosSe halló a 37 pacientes, lo que supone una incidencia aproximada de 1/10,000 recién nacidos vivos. Predominaba el sexo masculino (relación varón/mujer: 1,6/1). El tipo de AME diagnosticado más frecuentemente fue el tipo i (26 casos), seguido del tipo ii (9 casos), un caso de AME tipo iii, y otro caso de spinal muscular atrophy with respiratory distress type 1 (SMARD 1). La alteración genética más frecuente fue la deleción en homocigosis de exones 7 y 8 del gen SMN1, en 31 casos, mientras que 5 pacientes presentaban una genética atípica. La mediana de supervivencia para el tipo i fue de 8,0 meses y de 15,8 años para el tipo ii.
ConclusionesLa incidencia en nuestra población permanece estable en torno a 1/10,000. La mayoría de los casos presenta una genética típica con predominio de varones. En aproximadamente 1/10 pacientes la alteración genética fue diferente de la clásica. La prevalencia de AME no relacionadas con el gen SMN fue de 1/37. El nivel de cuidados se ha incrementado en los últimos años, en consonancia con las demandas sociales y asistenciales.
Our knowledge of spinal muscular atrophy (SMA) has advanced considerably since the earliest descriptions done by Werdnig, in 1891, and Hoffmann, in 1893. We know that it is due to a defect in the translation of the telomeric survival motor neuron (SMN) protein, which seems to play a role in several essential cellular functions (RNA metabolism, processing, and splicing) and other functions more specifically related to the survival of alpha motor neurons (apoptosis, axonal transport) in the anterior horn of the spinal cord. The SMN protein is encoded by the SMN1 and SMN2 genes. The literature describes a worldwide incidence of approximately 1 in 10,000 live births, with 1 out of 40 people being carriers of the disease.
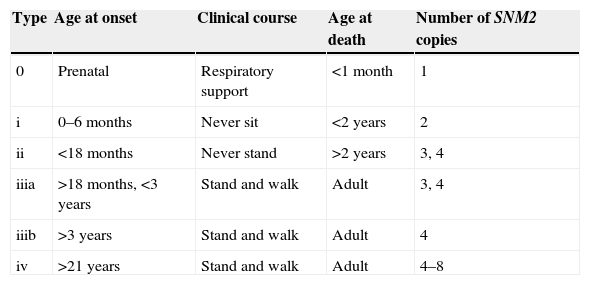
The classical clinical picture of SMA is predominantly proximal muscle weakness and atrophy, with ages of onset and severity that vary depending on the clinical type of SMA. Traditionally, the classification of SMA comprised 3 types: infantile, juvenile, and adult. At present, the International Consensus Statement for Standard Care in SMA1,2 classifies it into more types based on the age of onset and clinical course, dividing type III into subtypes according to the age of onset. Type IV was added for adult-onset cases, and type 0 for cases with prenatal onset, which result in death within the first weeks of life (Table 1). While there is clinical variability among individuals in each type and up to 25% of the patients cannot be categorised, this classification is useful for clinical practice and prognosis.
Classification of spinal muscular atrophy.
| Type | Age at onset | Clinical course | Age at death | Number of SNM2 copies |
|---|---|---|---|---|
| 0 | Prenatal | Respiratory support | <1 month | 1 |
| i | 0–6 months | Never sit | <2 years | 2 |
| ii | <18 months | Never stand | >2 years | 3, 4 |
| iiia | >18 months, <3 years | Stand and walk | Adult | 3, 4 |
| iiib | >3 years | Stand and walk | Adult | 4 |
| iv | >21 years | Stand and walk | Adult | 4–8 |
The gene involved in SMA, discovered in 1995,3 is located in the long arm of chromosome 5 (5q11.1-13.3). It has been named SMN for “survival motor neuron”. Humans have two nearly identical copies of this gene that have been called SMN1 and SMN2. They differ in a single nucleotide at the beginning of exon 7 (C in SMN1 and T in SMN2) that is important in the splicing of the SMN RNA. We do not know why this ubiquitous protein causes this highly selective neuronal disease. Deletions in SMN1 cause SMA. Approximately 95% of SMA patients have homozygous absence of exons 7 and 8 of the SMN1 gene, and about 5% are compound heterozygous for absence of exons 7 and 8 in one SMN1 allele and a point mutation in the other. Deletions in the SMN2 gene do not cause the disease; instead, it is the number of copies of this gene, which may vary, that has an effect on the phenotype and determines the severity of SMA, with a greater number of SMN2 copies correlating to a milder phenotype, although there are other factors at play.4,5 In this regard, it has been hypothesised that the disease could be treated with gene therapy by induced overexpression of the SMN2 gene, and the day when this terrible disease can be mitigated may not be far away.6,7Another genetic approach that was successful in an experimental study was the insertion of the SMN gene by means of viral vectors in the genome of a mouse model of the disease.8
Other forms of disease with clinical features that overlap with SMA, with subtle differences, are associated to different genes. Cases associated with chromosome 11 alterations have been described (spinal muscular atrophy with respiratory distress type 1 [SMARD1])9,10 as well as cases of early-onset recessive X-linked disease in males (Kennedy's disease).11,12
The aim of this study was to determine the present incidence of SMA in our population, its genotype distribution, and its epidemiological and clinical characteristics in the past 25 years in the context of our current knowledge, and to assess the level of care and the outcomes of SMA in our setting based on the international consensus.1,2
Materials and methodsWe performed a retrospective descriptive study by reviewing the clinical histories of patients with a clinical and neurophysiological diagnosis of spinal muscular atrophy assessed in our hospital in the past 25 years (from 1987 to early 2013).
We collected data for epidemiological, clinical and genetic variables, the supportive care received, and survival. The variables analysed were age at clinical onset and age at death, sex, family history of relatives that had the disease, initial symptoms, neurophysiological examinations, genetic testing, type of SMA, subsequent prenatal diagnostic examinations, supportive care received, and survival.
The molecular analysis involved amplification of exons 7 and 8 of the SMN1 gene by polymerase chain reaction from blood DNA samples from patients with suspected SMA. The SMN1 and SMN2 genes were differentiated by restriction fragment length polymorphism analysis. The absence of one of the 3 restriction fragments was interpreted as a homozygous deletion of exon 7 of the SMN1 gene. The homozygous deletion of exon 8 of the SMN1 gene was evinced by the absence of the 189-bp band of the SMN1 gene that was not digested in the presence of the 2 restriction fragments of the SMN2 gene.
In cases in which homozygous deletions were not detected, the number of copies of the SMN gene was quantified by multiplex ligation-dependent probe amplification. In patients with a single copy of the SMN1 gene (the other copy may be inactivated by a mutation) we sequenced the DNA of the coding part of the SMN1 gene to detect potential deletions, duplications and point mutations.
ResultsOverall epidemiological resultsWe found 37 patients who met the inclusion criteria, which amounts to an approximate incidence in our reference population and year of 1.41 cases per 15,000 live births (1 case per 10,638 live births). Fourteen patients were diagnosed between 1987 and 1999, while the number of diagnoses rose to 23 for the 2000 to 2013 interval, showing a clear increase in the number of diagnosed cases. Before 1995, cases were diagnosed by the identification of carriers in the family in 3 cases, and in other 3 the genetics of the patients were studied at a later point, as 2 of these patients had SMA type II and the other one SMA type III.
Most patients came from our reference area (31/37). The male sex predominated, with 23 cases in male patients and a 1.6 to 1 male to female ratio.
The mean age at the onset of symptoms was 2.2 months for type I SMA (range, 0–7 months) and 11.2 months for type II (range, 6–18 months). On the other hand, the only patient diagnosed with type III SMA was 12 months of age at the clinical onset, while the patient diagnosed with spinal muscular atrophy with respiratory distress (SMARD1) developed symptoms in the first month of life.
Clinical resultsThe most frequent type of SMA was type I, found in 26 patients, and followed by type II in 9. One case was diagnosed as type III SMA and another as SMARD1. There were no cases of X-linked inheritance (Fig. 1).

The presenting symptom in most patients was hypotonia, which occurred in 27 out of the 37 cases, followed by motor delay in 5 of the 37 cases. In 4 patients, type I SMA was associated with sucking and swallowing difficulties, and 2 of them had episodes of choking; another patient, who had type I SMA, was diagnosed following a life-threatening episode of apnoea. None of our patients had onset with arthrogryposis, nor they met the criteria for type 0 SMA. The presenting symptom in 3 patients was respiratory distress associated with intercurrent infection, and gait disorder in 2 patients (Fig. 2).

There were a total of 92 documented pregnancies in the families of the patients, which included 9 cases of spontaneous miscarriage and one case of voluntary termination of pregnancy. The nine miscarriages occurred in the families of 18 patients with type I SMA who were being followed up in our hospital (families in which there were a total of 47 pregnancies, corresponding to a 19.1% spontaneous miscarriage rate in this group).
Seven of the 37 patients had a family history of SMA, and 3 had a family history of idiopathic muscle weakness. In 4 patients there was a family history of neonatal death; 3 of these deaths occurred in the early neonatal period in siblings of one of the patients, who had onset with hypotonia at 8 days of age. The remaining 23 cases had no family history of neurological disease or associated symptoms (Fig. 3).
Genetic testing
Genetic testing was carried out in 36 patients. The number of copies of the SMN2 gene could not be determined in most patients. The most frequent genetic alteration was the homozygous deletion of exons 7 and 8 of the SMN1 gene, found in 31 out of the 36 cases, while 5 patients had atypical genetics. Four patients had compound heterozygous mutations with a classical deletion in one copy of the SMN1 gene and a non-functional truncated copy of the other resulting from different mutations: deletion of exons 1–6 in two cases, deletion of exon 1 in one case, and a 773insC mutation in the remaining case. Lastly, the patient diagnosed with SMA with respiratory distress had polymorphic changes in the IGHMPB2 gene of chromosome 11 (ex 1-T57C; ex 6-C714A and G823A; ex 8-C1104T; ex 9-A+18G; ex 13-A2011G and C2316T).
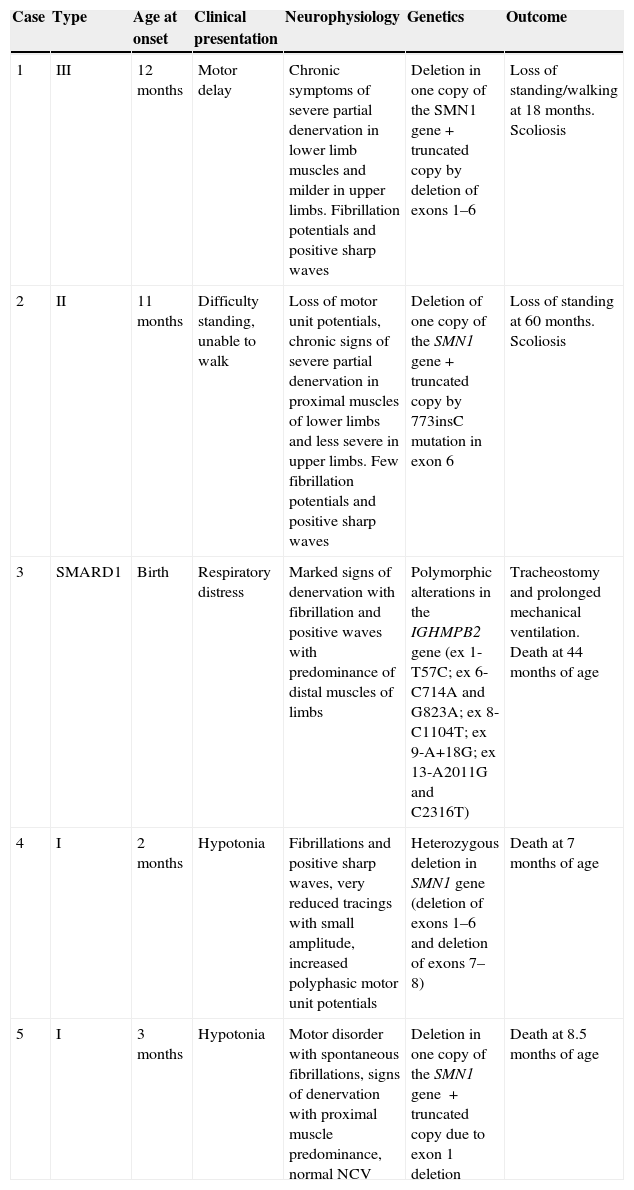
Table 2 summarises the epidemiological, clinical, neurophysiological and outcome characteristics of the 5 cases with atypical genetics. In six families, a genetic diagnosis was made prenatally in subsequent pregnancies (16.2%), leading to the identification of the disease in 4 pregnancies, carrier status in 2 pregnancies, and healthy non-carrier status in the remaining 2 pregnancies (Fig. 4).
Epidemiological, clinical, neurophysiological and clinical course characteristics of atypical genetics cases.
| Case | Type | Age at onset | Clinical presentation | Neurophysiology | Genetics | Outcome |
|---|---|---|---|---|---|---|
| 1 | III | 12 months | Motor delay | Chronic symptoms of severe partial denervation in lower limb muscles and milder in upper limbs. Fibrillation potentials and positive sharp waves | Deletion in one copy of the SMN1 gene+truncated copy by deletion of exons 1–6 | Loss of standing/walking at 18 months. Scoliosis |
| 2 | II | 11 months | Difficulty standing, unable to walk | Loss of motor unit potentials, chronic signs of severe partial denervation in proximal muscles of lower limbs and less severe in upper limbs. Few fibrillation potentials and positive sharp waves | Deletion of one copy of the SMN1 gene+truncated copy by 773insC mutation in exon 6 | Loss of standing at 60 months. Scoliosis |
| 3 | SMARD1 | Birth | Respiratory distress | Marked signs of denervation with fibrillation and positive waves with predominance of distal muscles of limbs | Polymorphic alterations in the IGHMPB2 gene (ex 1-T57C; ex 6-C714A and G823A; ex 8-C1104T; ex 9-A+18G; ex 13-A2011G and C2316T) | Tracheostomy and prolonged mechanical ventilation. Death at 44 months of age |
| 4 | I | 2 months | Hypotonia | Fibrillations and positive sharp waves, very reduced tracings with small amplitude, increased polyphasic motor unit potentials | Heterozygous deletion in SMN1 gene (deletion of exons 1–6 and deletion of exons 7–8) | Death at 7 months of age |
| 5 | I | 3 months | Hypotonia | Motor disorder with spontaneous fibrillations, signs of denervation with proximal muscle predominance, normal NCV | Deletion in one copy of the SMN1 gene +truncated copy due to exon 1 deletion | Death at 8.5 months of age |

Twenty-five patients were followed up at our hospital, 7 of them through at-home hospital care. Most of them had SMA type I (18 patients). The supportive care they received included motor physical therapy and rehabilitation (15 patients), respiratory physical therapy (8 patients) or basic respiratory care such as secretion management and aspiration (5 patients). Two patients received at-home oxygen therapy through low-flow nasal cannulae at 6 and 7 months, and noninvasive ventilation was initiated in one patient at 8 months. Two patients were treated with invasive mechanical ventilation with tracheostomy. One of them was the patient diagnosed with SMARD1, in whom invasive ventilatory support was initiated before the diagnosis was confirmed. The second one was a SMA type II patient in whom a proactive intervention was performed in agreement with the family after prolonged invasive mechanical ventilation; the tracheostomy was performed at age 12 months, and after discharge from the hospital at 4 years of age the follow-up consisted of at-home hospital care until the patient's death at 14 years of age.
As for nutritional management, 4 patients required enteral nutrition, delivered at home through a nasogastric tube, and a gastrostomy was performed in one patient (Fig. 5). The parents of one other patient refused to have a gastrostomy performed.
, invasive mechanical ventilation (IMV), enteral nutrition through nasogastric tube (NGT) or gastrostomy.")
Supportive care received by patients followed up in our centre. Motor rehabilitation, respiratory physical therapy, basic respiratory care, low-flow oxygen therapy, noninvasive mechanical ventilation (NIMV), invasive mechanical ventilation (IMV), enteral nutrition through nasogastric tube (NGT) or gastrostomy.
We know that 18 out of the total 25 patients followed up in our hospital died, and most of them had SMA type I (only one patient with SMA type II died during this period). The median survival in our series for type I patients was 8.0 months (range, 1.5–44 months) and 15.8 years for type II (range, 3–25.8 years). An important fact is that 22 of the 25 patients with SMA type I followed up died before 24 months of age.
DiscussionAt the end of the 25 years under study, the incidence in the reference population and year was similar to those described in other series, which suggests a stable genetic penetrance for this disease in our population.13 SMA type I accounted for 70.3% of the cases; the genetic defect in most of these patients (31/36) was a homozygous deletion in the SMN1 gene; the mean survival was less than one year for type I, somewhat below what is reported in the literature; and there was a predominance of male patients in our series.
Hypotonia in infancy continues to be the most common clinical presentation (Fig. 2), although in some cases (3 in our series) the presenting symptom is respiratory distress. The age of clinical onset in patients with SMA type I was 2.2 months. While the diagnosis was made relatively early, at present it is believed that this is not early enough to achieve the benefits expected of emerging therapeutic approaches.14–17 The usefulness of neonatal screening for offering specific patients participation in clinical trials is being considered. This approach has already been implemented and there are studies on the subject.18
We found a spontaneous miscarriage rate of 19.1% in the families of patients with SMA type I, at the upper limit of the range described for the general population (10–20%). However, this figure may underestimate the actual rate of spontaneous miscarriage associated with this disease, as we have no information on the occurrence of subsequent miscarriages in families that were not followed up in our hospital. Although this has been described before, there are very few published studies on the association of SMA with spontaneous miscarriage, and we did not find any systematic reviews on this matter. A published case associated foetal death to foetal akinesia-deformation sequence.19
Only one of our patients had a form of SMA unrelated to the SMN gene. The patient was diagnosed with SMARD1. The polymorphisms found in this patient had never been described before. We assumed they were involved in the aetiology based on their location in chromosome 11 and the patient's clinical features. The management of this case involved early ventilatory support with tracheostomy due to its onset with severe respiratory distress for which no initial clinical diagnosis was made, as the patient followed the natural course of this disease in its early stages with severe respiratory problems and predominantly distal involvement.20 Several publications debate the use of invasive respiratory support in SMA type I, which poses an ethical dilemma that has yet to be resolved.21 Since 2007 there is a standard of care consensus22 that aims to improve the quality of life of SMA patients, although according to several studies in different countries its introduction has raised controversy.23
In our experience, implementing this standard of care, as we have done in recent years, requires the coordinated effort of the various specialties involved, as well as adequate development of at-home hospital care. It requires, at least, the universalisation of at-home oxygen therapy, gastrostomy and noninvasive ventilatory support during the course of the disease, in addition to the optimisation of orthopaedic measures to prevent bizarre deformities, while adjusting care to the wishes and expectations of the parents. It remains to be seen whether this approach increases survival, and while this is not the goal of the care, it could have this logical result. Patient associations support this standard of care, which must be taken into account by paediatricians. The median survival for type I patients was 8 months, lower than the figures published to date, which was probably due to our late introduction of active nutritional and respiratory support. This precludes us from drawing conclusions about overall survival, although this approach seems to alleviate the anxiety of parents in regards to the suffering of their child.
In our series, tracheostomy was performed in only two patients. One was the patient with SMARD1, who had it for the reasons stated above, and the other one a patient with SMA type II, in whom it was more likely to be indicated given the slower course of the disease. Nutritional support was only provided to one patient, after the parents agreed, through a scheduled gastrostomy at 16 months of age. A cough assist machine was used in two patients with SMA type I at 8 and 23 months of age, resulting in different degrees of satisfaction, as in one case it worsened the patient's condition and the parents decided to discontinue its use.
The genetic mutations found in our series are similar to those reported in the literature (Table 2). In 2011, the only 2 families in which the truncated copy of the SMN1 gene with the deletion of exons 1–6 was detected came from the same geographical region, and in both cases the copy was inherited from the father. Analysis of the polymorphic markers linked to the SMN1 locus and of multilocus markers (272 and 212) revealed that the fathers of the two patients had the same haplotype, which suggested that both chromosomes shared a common ancestor.
Conclusions and commentsTo conclude, our series shows a classical clinical course with a relatively early onset and a stable population incidence of approximately 1 case per 10,000 live births per year. Most patients had a typical genetic basis, and there was a predominance of males. Approximately 1 in 10 patients had a mutation other than the classical mutation in the SMN gene. Only one of the 37 patients in our series had a mutation in a gene other than the SMN gene that corresponded to a case of SMARD1. The overall survival in our series was below that described previously. The level of care has increased in line with social and welfare demands in recent years.
We believe that multicentre cooperation is needed to adequately address the research and management of this disease, for which there is an increasingly hopeful future.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors want to thank Dr. Carmen de Benito and Dr. Emilia del Castillo, Servicio de Genética, HRU Carlos Haya.
Please cite this article as: Rodríguez AM, Martínez PLM, Fernández JMR, Cardona AU, Antón JM. Atrofia muscular espinal: revisión de nuestra casuística en los últimos 25 años. An Pediatr (Barc). 2015;82:159–165.
