
The parietal foramina are small openings measuring approximately 1 mm near the sagittal border of the parietal bones. They are a normal variant, unilateral or bilateral, found in up to 60% of the population. The term giant parietal foramina or foramina parietalia permagna (FPP) refers to openings in this region with a diameter greater than 5 mm, and its incidence is much lower (1 per 15,000–25,000 individuals).1 In fact, this denomination is inaccurate, as small parietal foramina may coexist with giant foramina, and these large openings are not always an enlarged parietal foramen.
Foramina parietalia permagna is a primary ossification defect at the level of the parietal eminence. In young children, it may present as a persistently enlarged posterior fontanelle, with subsequent ossification of a midline bridge during childhood giving rise to the two foramina.1
In most cases, FPP is asymptomatic, although it may be associated with headache, nausea, vomiting or intellectual disability and sometimes to other malformations: craniosynostosis, cortical dysplasia, microcephaly, ear or eye abnormalities, craniofacial dysostosis, syndactyly, multiple exostosis or cerebrovascular abnormalities.1,2
Eighty-five percent of cases are due to genetic variants with an autosomal dominant pattern of inheritance and a high but incomplete penetrance (90%).1 Most of the variants described to date have been in the MSX2 (5q35.2) or ALX4 (11p11.2) genes. The prevalence of variants in each of these genes is similar, and the clinical presentations are nearly indistinguishable, since both MSX2 and ALX4 are homeobox genes that encode homeodomain transcription factors involved in skeletal development.3
The diagnosis of FPP is based on the family history and physical examination, with the possible addition of imaging tests: the chest radiograph features symmetric lucencies in the parasagittal zone of parietal bones, computed tomography (CT) with 3D reconstruction can demarcate the bony defect and magnetic resonance imaging (MRI) can detect other associated intracranial abnormalities.1 The diagnosis is confirmed by the detection of a pathogenic variant using molecular techniques.
Treatment is usually conservative, with options including preventive measures such as cranial orthosis to avoid brain injury. Surgery is reserved for large foramina associated with a high risk of injury: young children who are very active, have seizures or with additional malformations requiring surgery.1,4
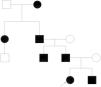
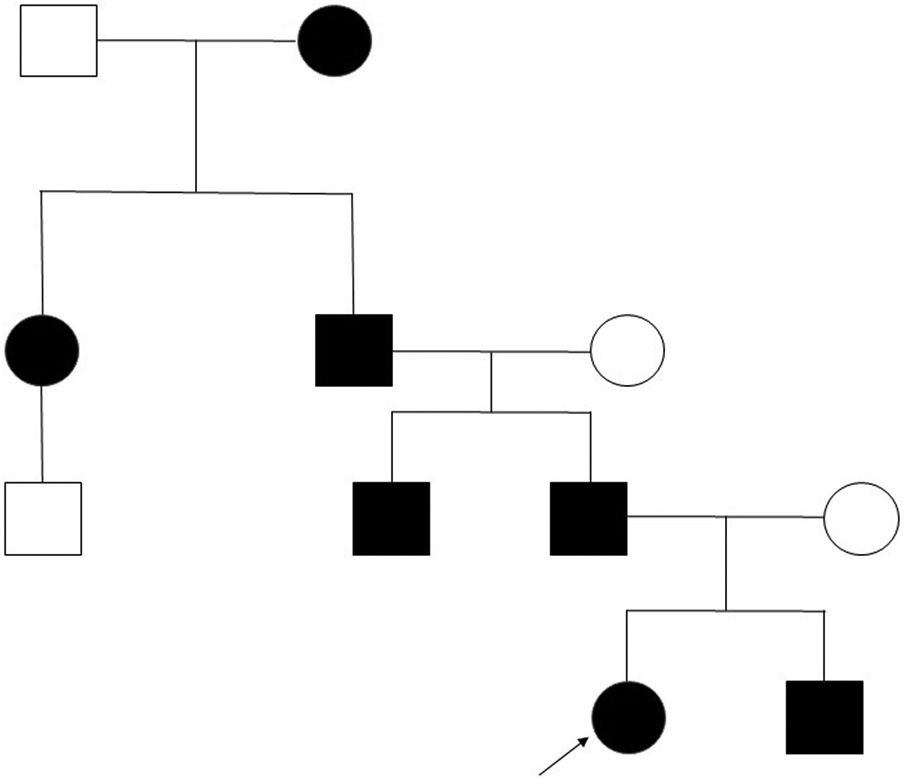
Clinical case. We present the pedigree of a Spanish family with 7 members in 4 consecutive generations (Fig. 1) with a diagnosis of FPP that carried a heterozygous novel variant in the ALX4 gene (c.404_405dupG).
The index case was a newborn girl with a diagnosis of cranial bone defects made by ultrasound in the prenatal period that led to scheduling an elective caesarean delivery. At birth, the infant had an enlarged posterior fontanelle (5 × 5 cm) in the absence of additional craniofacial anomalies, with a normal appearance and a normal neurologic examination. A transfontanellar ultrasound examination ruled out additional malformations, but also detected bilateral echogenic lesions in the parietooccipital region compatible with infarction possibly secondary to trauma, confirmed by means of a cranial MRI (Fig. 2A). At 3 years of follow-up, the patient remains asymptomatic, with normal neurodevelopment.
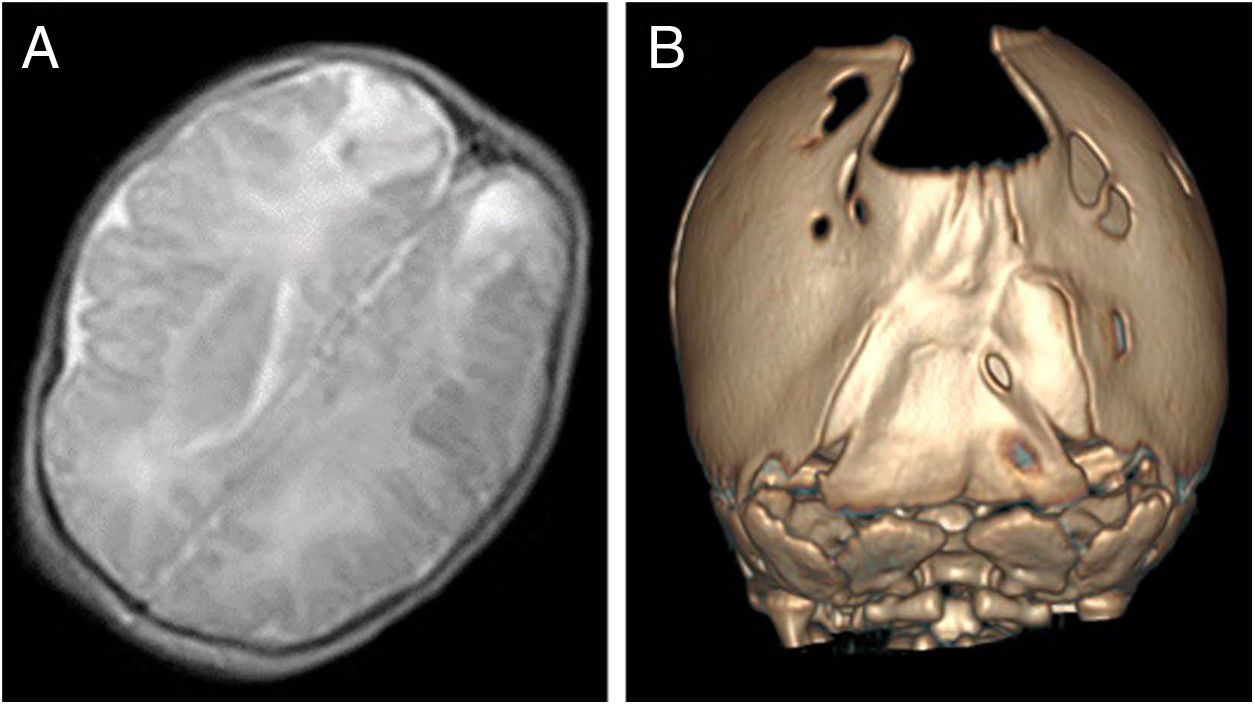
In a younger brother, the diagnosis was made antenatally by MRI, which revealed absence of bone tissue in the parieto-occipital sulcus. In the initial visit to the paediatric neurology unit, the examination revealed frontal bossing with occipital flattening and bone crests in both lambdoid sutures and the parieto-occipital defect on palpation. A CT scan of the calvarium was performed, leading to diagnosis of bilambdoid synostosis associated with FPP (Fig. 2B). The findings of the neurologic examination were normal. Surgery was performed to correct the craniosynostosis.
The only significant finding in the evaluation of the father was the presence of the parietal openings characteristic of FPP, in the absence of any other manifestation, as was also the case in the other 4 affected members of the family.
Foramina parietalia permagna is a rare and benign disorder. Knowledge of its clinical and genetic characteristics can prevent performance of unnecessary tests and treatments, reducing the anxiety of parents, especially in case of a prenatal diagnosis. When bilateral parietal openings are detected on prenatal ultrasound examinations, investigation of the pedigree is useful for the purpose of identifying other cases in the family that may have gone undetected due to intrafamilial variability or spontaneous closure of the bone defect. The diagnosis can be confirmed through the clinical and radiological evaluation and the detection of changes in the ALX4 or MSX2 genes in relatives or in the foetus.5,6 The presence of a skull defect in and of itself is not an indication for elective caesarean delivery since, as occurred in the case presented in this article, this intervention does not completely eliminate the risk of trauma secondary to the birth process.
Please cite this article as: Bote Gascón M, Martínez del Río C, García Ron A. Foramina parietalia permagna: evaluación clínico radiológica de una familia española con mutación no descrita en el gen ALX4 (c.404_405dupG). An Pediatr (Barc). 2021;95:121–122.
Previous presentation: this case was presented as a poster at the 66th Congress of the Asociación Española de Pediatría, June 7–9, 2018, Zaragoza, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals