

Biallelic mutations in the OTUD6B gene, which is located in region 8q21.3, have been recently described as causing syndromic intellectual disability (ID) in 7 families across the world.1,2 This gene encodes an enzyme involved in deubiquitination, the removal of ubiquitin from proteins marked for degradation. Ubiquitinating/deubiquitinating enzymes also regulate numerous processes, such as cell signalling, protein–protein interactions and intracellular trafficking.3 Recent studies have found an association of changes in these mechanisms with autoinflammatory4 and neurologic5 disorders, among other diseases, and changes in the gene encoding deubiquitinase OTUD6B with abnormalities in cell growth and cancer.6
Intellectual developmental disorder with dysmorphic facies, seizures, and distal limb anomalies OMIM 617452 manifests with all the characteristics that define it in every case described to date, and the ID is usually severe. Other features frequently found in these patients are a history of intrauterine growth restriction IUGR, short stature, heart defects and various skeletal abnormalities and neurologic disorders autism spectrum disorder, ataxia, etc. On account of its exceptional nature, we describe a new case with a homozygous c.433C > T mutation in gene OTUD6B previously detected in 3 of the 7 families described in the literature.
The patient was a Spanish girl aged 4 years with no relevant family history born to healthy nonconsanguineous parents. She was referred to the medical genetics clinic at age 3 months due to the presence of abnormal facial features, microcephaly and a history of IUGR. The pregnancy developed without complications until week 30, when imaging detected the absence of the nasal bone and suggested the presence of a ventricular septal defect (unconfirmed). An amniocentesis was performed, and the results of quantitative fluorescence PCR and karyotyping were normal. The patient had no perinatal disease and her anthropometric values were normal (weight, 2745 g [15th percentile, z = −1.05]; length, 48 cm [21st percentile; z = −0.83]; head circumference [HC], 33 cm [19th percentile, z = −0.9]).
During the follow-up, the patient exhibited generalised hypotonia and moderate global developmental delay. The patient achieved sitting up at 11 months and currently exhibits unstable walking. She has features of autism spectrum disorder and has expressive language delay. She had onset of febrile tonic-clonic seizures at age 8 months that have since become afebrile. The findings of electroencephalography (EEG) and video-EEG examinations have been normal to present. Nevertheless, treatment with valproic acid was initiated, which has achieved adequate control of the seizures. The patient is enrolled in an early intervention programme and is in follow-up with periodic evaluations in the paediatric neurology department. Magnetic resonance imaging revealed a mild and nonspecific enlargement of the fourth ventricle, in the absence of other CNS anomalies.
In the early months of life, the patient developed a moderate gastro-oesophageal reflux that required treatment with omeprazole, accompanied by difficulty swallowing. At present, she consumes all food in pureed form. She has not required surgeries or nasogastric tube feeding. The patient also experiences constipation, which has been managed successfully with dietary measures.
As for growth, she has short stature (95 cm; 2nd percentile, z = −2.1) and moderate microcephalia (HC, 45 cm; below 1st percentile, z = −4.21). During the follow-up, we screened for other potential congenital anomalies, with detection of horseshoe kidney leading to ongoing follow-up in the nephrology department. Renal function is normal.
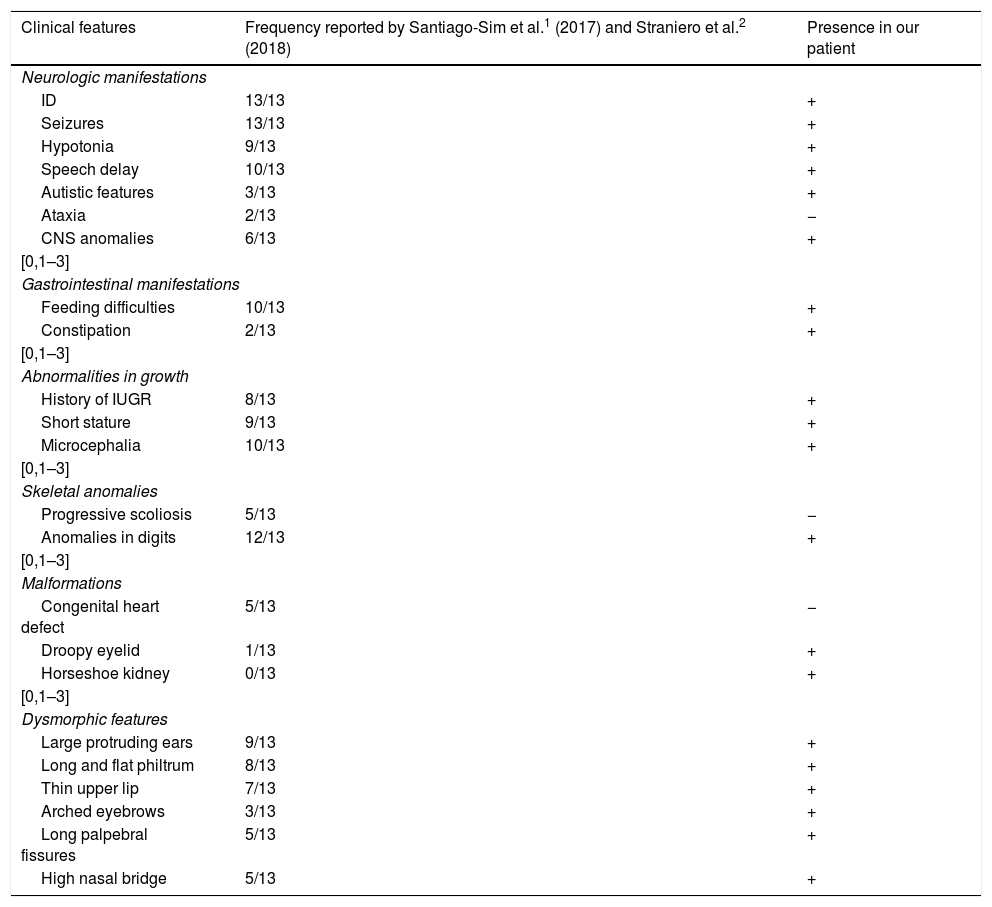
The patient has also been assessed in the ophthalmology department due to left eyelid ptosis accompanied by mild strabismus, leading to diagnosis of myopia, for which the patient wears corrective lenses. Hearing is normal. The skeletal features include a short neck, postural kyphosis, flat feet with a prominent heel, limited range of extension in the elbows and knees and digit malformation consisting of a widening of the distal phalanges, bilateral clinodactyly in the little finger, thumb, and first and fifth toes, superimposition of the little finger over the ring finger and bilateral shortening of the second toe. Table 1 compares the features found in our patient compared to features described in the previous literature.
Clinical characteristics of the cases described in the literature.
| Clinical features | Frequency reported by Santiago-Sim et al.1 (2017) and Straniero et al.2 (2018) | Presence in our patient |
|---|---|---|
| Neurologic manifestations | ||
| ID | 13/13 | + |
| Seizures | 13/13 | + |
| Hypotonia | 9/13 | + |
| Speech delay | 10/13 | + |
| Autistic features | 3/13 | + |
| Ataxia | 2/13 | − |
| CNS anomalies | 6/13 | + |
| [0,1–3] | ||
| Gastrointestinal manifestations | ||
| Feeding difficulties | 10/13 | + |
| Constipation | 2/13 | + |
| [0,1–3] | ||
| Abnormalities in growth | ||
| History of IUGR | 8/13 | + |
| Short stature | 9/13 | + |
| Microcephalia | 10/13 | + |
| [0,1–3] | ||
| Skeletal anomalies | ||
| Progressive scoliosis | 5/13 | − |
| Anomalies in digits | 12/13 | + |
| [0,1–3] | ||
| Malformations | ||
| Congenital heart defect | 5/13 | − |
| Droopy eyelid | 1/13 | + |
| Horseshoe kidney | 0/13 | + |
| [0,1–3] | ||
| Dysmorphic features | ||
| Large protruding ears | 9/13 | + |
| Long and flat philtrum | 8/13 | + |
| Thin upper lip | 7/13 | + |
| Arched eyebrows | 3/13 | + |
| Long palpebral fissures | 5/13 | + |
| High nasal bridge | 5/13 | + |
CNS, central nervous system; ID, intellectual disability; IUGR, intrauterine growth restriction.
The patient had dysmorphic features that evolved with growth, as can be seen in Fig. 1. The parents signed an informed consent form allowing publication of these photographs.

After the initial evaluation, we ordered a 60 K oligonucleotide array-based comparative genomic hybridisation analysis, the results of which were normal. Due to the suspicion of Nicolaides–Baraitser syndrome, Baraitser–Winter syndrome and cerebrofrontofacial syndrome, additional genetic tests were performed with next generation sequencing methods, revealing no mutations. Eventually, performance of whole exome sequencing identified a previously described c.433C > T homozygous c.433C > T mutation in the OTUD6B gene,1 confirming that the parents were carriers of the variant.
In short, we have described the first case in Spain, with features that are consistent with previous descriptions and with additional phenotypic manifestations, as the patient also had horseshoe kidney and myopia.
This new case supports the proposed role of the OTUD6B gene in syndromic ID and once again demonstrates the considerable usefulness of exome sequencing for diagnosis of phenotypes that are complex, have been described only recently and are extremely infrequent whose manifestations overlap with those of classic syndromes and that are probably underdiagnosed due to a lack of knowledge.
The variant was the same as the one identified in 3 of the 7 families described in the literature, which suggests that this is a recurrent mutation.
The differential diagnosis must include congenital disorders of glycosylation, Nicolaides–Baraitser syndrome and Baraitser–Winter syndrome as well as the aforementioned Rubinstein–Taybi syndrome and Kabuki syndrome.
Please cite this article as: Sánchez-Soler MJ, et al. Primer caso español de discapacidad intelectual sindrómica con dismorfia facial, crisis y anomalías de extremidades por mutaciones bialélicas en el gen OTUD6B. An Pediatr (Barc). 2020;92:169–171.
