









STXBP1 syndrome is a genetic disorder that affects one of the regulatory mechanisms of neurotransmitter release by the synaptic vesicles and has serious implications for neurodevelopment. Symptoms usually appear in the first days or months of life, and very often include epilepsy, psychomotor delay, and intellectual disability. Although it was initially regarded as an early epileptic encephalopathy, the increase in the number of cases diagnosed, as well as the advances in research have been expanding the phenotype and characterising this disease as a disorder of neurodevelopment. Furthermore, on being linked to epileptic problems, this genetic mutation could be associated with many cases of intellectual disability and movement disorders of unknown cause.
ObjectivesTo describe the characteristics of the patients identified in Spain with STXBP1 syndrome, and the implications for the diagnosis of these characteristics.
Patients and methodsThe details are presented on 17 individuals, aged between 2 years and 17 years, diagnosed in Spain with STXBP1 syndrome.
ConclusionsThere is a clear under-diagnosis of STXBP1 syndrome in Spain. Besides the inherent diversity of the disorder, with the increase in the number diagnoses the variability of the phenotype is even wider. The description of the alarm signs is necessary in order to identify those individuals with less prototypical manifestations of the disorder
El síndrome STXBP1 es una enfermedad genética que afecta a uno de los mecanismos reguladores de la liberación de neurotransmisores por parte de las vesículas sinápticas, por lo que tiene serias implicaciones para el neurodesarrollo. Suele manifestarse en los primeros días o meses de vida, e incluye con mucha frecuencia epilepsia, retraso psicomotor y discapacidad intelectual. Aunque inicialmente se consideró una encefalopatía epiléptica precoz, el aumento de los casos diagnosticados y el avance de la investigación ha ido ampliando el fenotipo y caracterizando esta enfermedad como un trastorno del neurodesarrollo. Además de estar asociada a alteraciones epilépticas, esta mutación genética puede estar asociada a muchos casos de discapacidad intelectual y trastornos del movimiento cuya causa se desconoce.
ObjetivosDescribir las características de los pacientes identificados en España con el síndrome STXBP1, y las implicaciones para el diagnóstico de dichas características.
Pacientes y métodosSe presentan los datos de 17 personas diagnosticadas en España con el síndrome STXBP1 de edades comprendidas entre los 2 y los 17 años.
ConclusionesExiste un claro infradiagnóstico del síndrome STXBP1 en nuestro país. Además de la diversidad inherente al trastorno, con el aumento del número de diagnósticos, la variabilidad fenotípica se amplía aún más. Se hace por tanto necesaria la descripción de signos de alarma que permitan identificar a aquellas personas con manifestaciones menos prototípicas de la alteración.
STXBP1 syndrome is a genetic disorder with a significant impact on neurodevelopment. The STXBP1 gene is involved in the regulation of exocytosis processes, that is, with the mechanisms that control the synapse. More specifically, it is involved in neurotransmission. The syntaxin binding protein, also known as Munc18-1, binds syntaxin to form a complex that regulates the release of neurotransmitters from synaptic vesicles.1–3
Mutations in the STXBP1 gene were first described in 2008 in patients with Ohtahara syndrome, also known as early infantile epileptic encephalopathy.4 The clinical manifestations described in these patients included very early onset epilepsy, motor deficits and psychomotor retardation. The STXBP1 gene has also been associated with various epileptic syndromes, such as West syndrome, malignant migrating partial seizures of infancy, Dravet syndrome, severe early myoclonic encephalopathy, Rett syndrome with or without seizures, the combination of autism and epilepsy or epileptic dyskinetic encephalopathy.5 With the advances in genetic testing, the clinical spectrum of this gene has been progressively widening. In a sample of children with early infantile epileptic encephalopathy without Ohtahara or West syndrome, Deprez et al.6 found mutations in the STXBP1 gene in 10% of the patients. Also, mutations in this gene have been found in a significantly higher proportion of individuals with autism spectrum disorder (ASD) compared to the general population.7
Although epilepsy seemed a defining characteristic of the syndrome, there has been an increasing number of cases reported in the literature of mutations in the STXBP1 gene in individuals that never had seizures. Hamdan et al.8 analysed a sample of patients with nonsyndromic intellectual disability and found a mutation in the STXBP1 gene in 1 individual aged 21 years that had never had epilepsy. Later on, there have been other reports of patients with movement disorders and intellectual disability that had mutations in the STXBP1 gene in the absence of epilepsy,9 which has further broadened the clinical picture of STXBP1 syndrome. Although mutations in this gene were initially considered one of the main causes of early-onset epileptic encephalopathy, subsequent studies have demonstrated that epilepsy is just one manifestation within a broad constellation of symptoms with a heterogeneous expression. Thus, STXBP1 syndrome is a neurodevelopmental disorder rather than an epileptic encephalopathy.10–12
The differential diagnosis must include other diseases associated with epileptic features with early onset, such as pyridoxine-dependent epilepsy, pyridoxamine 5′- phosphate oxidase (PNPO) deficiency, cerebral folate deficiency or glucose transporter type 1 (GLUT1) deficiency.13
Descriptions of the clinical presentation of individuals with STXBP1 syndrome suggest that when epilepsy is present, it has a very early onset. The mean age at onset of seizures is 6 weeks post birth, and the range goes from the first day of life to 12 years.10 Controlling the seizures is usually challenging at the beginning, although outcomes on this aspect are quite heterogeneous. Published studies are consistent in reporting a favourable outcome in most individuals with mutations in this gene, although a small proportion continue to have frequent seizures.4–6,12 Stamberger et al.10 found that in a sample of 147 patients, nearly half (43.8%) stopped experiencing seizures between 1 month and 4 years of age, with the median age at resolution of 8 months. However, 28.7% of the sample continued to have frequent seizures (more than once a week despite treatment). These authors also found recurrence of epilepsy after a long period of remission in 6 of the patients under study. All patients had intellectual disability, generally requiring considerable support. Cognitive impairment was limited to learning disabilities in only 1 case.
Nearly half of the individuals identified by Stamberger et al.10 were able to take a few steps independently. They developed this skill between ages 14 months and 6 years. Fifteen percent of the patients had some ability to communicate verbally, and ASD was present in 17% of the sample. The authors also described a high frequency of movement disorders, such as hypotonia, ataxia, tremors, spasticity, dyskinesia and dystonia.10
The variability in the phenotype of STXBP1 disorders may lead to their underdiagnosis.11 The analysis of a birth cohort spanning 10 years in Denmark found a prevalence of 1/91 862.10 To date, there are no data on the prevalence of STXBP1 syndrome in Spain, but based on the current literature it appears that this syndrome may be associated with a considerable number of neurodevelopmental disorders manifesting with movement problems and epileptic features.
The aim of our study was to describe the characteristics of the population of individuals known to have STXBP1 syndrome in Spain and their diagnostic needs. The description of the features of this syndrome and its early clinical manifestations may help identify new cases and reduce the delay in diagnosis.
Patients and methodsAt the beginning of the study period, there were 20 individuals in Spain diagnosed with STXBP1 syndrome that had made contact with the STXBP1 Association, either of their own initiative or after being referred by the department that made the diagnosis. We sent the families information about the study as well as an informed consent form in case they chose to participate. The information presented in this article has been drawn from a larger study whose aim is to describe the characteristics of this collective in Spain, define its needs and propose priority guidelines to improve the quality of life of affected individuals and their families. Of the 20 families that we invited to participate, 17 accepted (85%). Once we obtained the informed consent forms, we requested information from the families through 3 online questionnaires sent at 3 different time points. These questionnaires were completed on an anonymous basis by one or both parents. The questionnaires collected information on the individual with STXBP1 syndrome, the characteristics of the family, and the personal experiences and perception of different processes and aspects of life as an individual with STXBP1 syndrome or a family member.
ResultsGeographical distributionFour families from Barcelona, 3 from Madrid, 2 from Valencia and 1 each from Alicante, Toledo, Jaen, Valladolid, Zaragoza, Malaga, Leon and Avila participated in the study at different points.
Table 1 presents the estimated frequency of cases calculated by extrapolation of the prevalence data published by Stamberger et al.10 to the Spanish population aged less than 19 years based on census data obtained by the Instituto Nacional de Estadística (National Institute of Statistics, INE) in 2017 and the actual number of identified cases for each autonomous community.
Estimated frequency of cases in the Spanish population and actual number of detected cases by autonomous community.
| Autonomous community | Estimated frequency in <19 years | Actual number of detected cases |
|---|---|---|
| Andalusia | 20 | 2 |
| Aragon | 3 | 1 |
| Asturias | 2 | 0 |
| Balearic Islands | 3 | 0 |
| Canary Islands | 4 | 0 |
| Cantabria | 1 | 0 |
| Castilla y Leon | 4 | 4 |
| Castilla-La Mancha | 5 | 1 |
| Catalonia | 17 | 4 |
| Valencia | 11 | 3 |
| Extremadura | 2 | 0 |
| Galicia | 5 | 0 |
| Madrid | 15 | 3 |
| Murcia | 4 | 0 |
| Navarre | 1 | 0 |
| Basque Country | 4 | 0 |
| La Rioja | 1 | 0 |
| Ceuta | 0 | 0 |
| Melilla | 0 | 0 |
| Total | 101 | 17 |
Based on the data published by the INE, there are approximately 400 000 births per year in Spain, which would correspond to about 4 new cases per year in our country.
Characteristics of individuals with STXBP1 syndromeSex and ageOf the 17 participants in the study, 7 were female (41%) and 10 male (59%). The age range was 2–17 years. The mean age was 8.3 years, the standard deviation 4.7 years and the median 7 years.
EpilepsyOne of the main features in individuals with STXBP1 syndrome and that is frequently the first manifestation indicative of a disorder is the development of seizures. As can be seen in Fig. 1, the individuals in our sample followed a distribution that is consistent with the current literature on the subject.
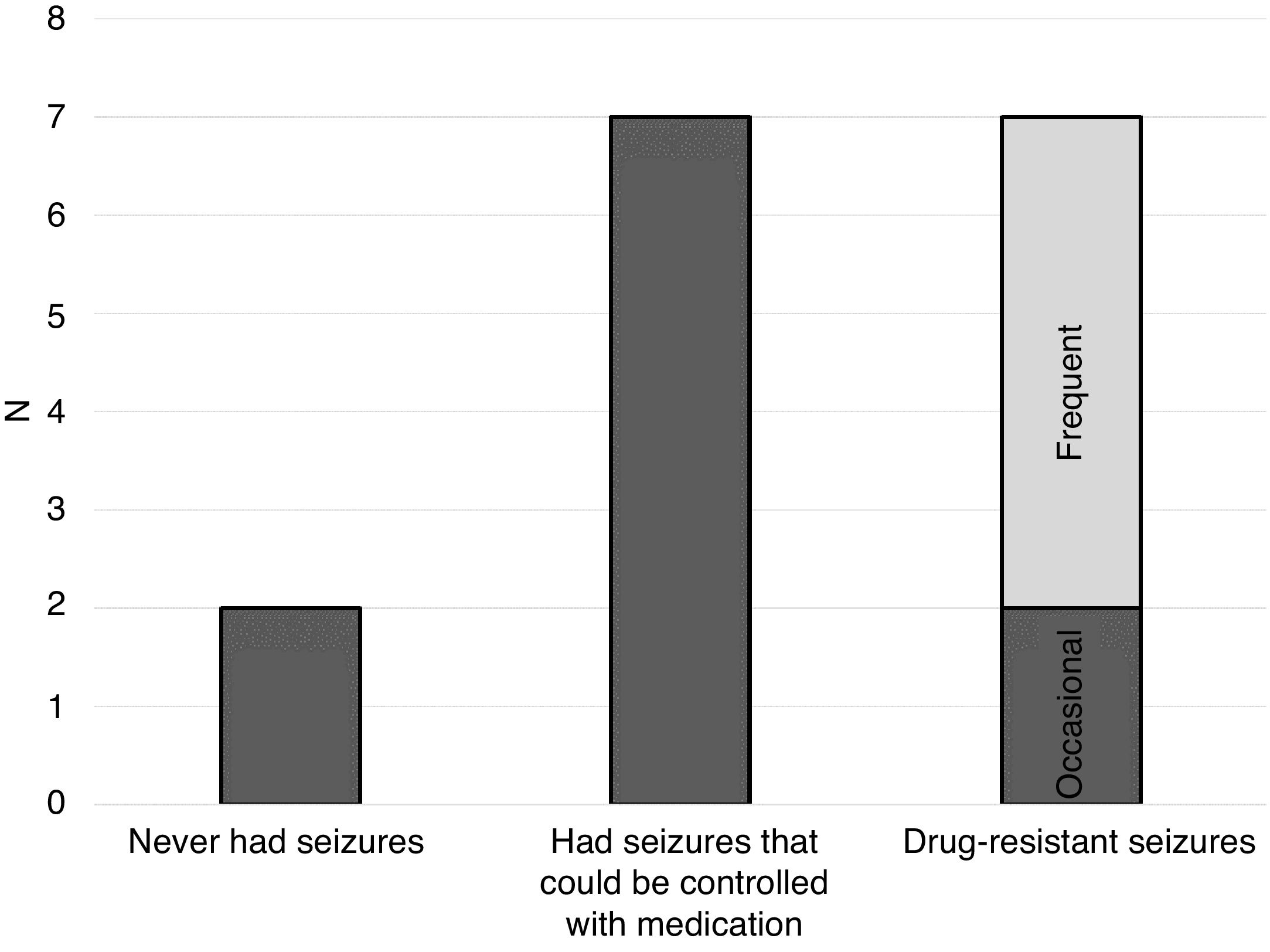
Most of the individuals with STXBP1 syndrome in our sample have had seizures at some point (46%), but they could be controlled with medication; 13% have seizures occasionally (less than once a week) and 33% have frequent seizures (weekly or more frequently). One participant has never had epileptic seizures. Another participant has never had a seizure, but has exhibited abnormal electroencephalographic activity, and the family has been informed of the presence of “epileptic peaks”.
The onset of epilepsy usually occurs at a very early age. In our sample, the age of onset ranged from day 1 post birth to 2 years and 9 months. The median age of onset was 7 days. As for the type of epilepsy, we found a wide spectrum of forms of epilepsy in our sample consistent with the previous literature. Table 2 shows the percentage of participants that have presented with each form of epilepsy. The most frequent presentations were epileptic spasms and tonic-clonic seizures.
Types of seizures found in participants.
| Forms of epilepsy | % |
|---|---|
| Epileptic spasms | 71.4 |
| Generalised seizures: tonic-clonic seizures | 71.4 |
| Focal aware seizures | 50 |
| Generalised seizures: absence seizures | 42.8 |
| Generalised myoclonic seizures | 42.8 |
| Focal to bilateral seizures | 28.5 |
| Focal impaired awareness seizures | 28.5 |
| Generalised atonic seizures | 14.2 |
When it came to pharmacological treatment, in all cases more than one drug was tried until control of seizures was accomplished. Three of the participants are also following a ketogenic diet.
Mutations in the STXBP1 gene are sometimes associated with other syndromes that manifest with seizures; in our sample, 1 participant had a diagnosis of West syndrome, another a diagnosis of Ohtahara syndrome, 2 have diagnoses of both syndromes (West and Ohtahara) and, lastly, 1 has diagnoses of West, Ohtahara and Dravet syndromes.
Identified disability levelAll individuals that participated in the study had some degree of disability that was officially established, ranging from a level of 40% to levels exceeding 90%, and thus needed different levels of support.
Motor developmentIn terms of motor development, individuals with STXBP1 syndrome exhibit considerable variability in their ability to move independently. We obtained data from 15 families regarding this aspect. We found that 60% of affected individuals were able to move independently to some degree, either in specific settings or with assistance, or to travel small distances in the immediate environment, while 40% were at the time of the study unable to move independently at all. Fig. 2 shows the degrees to which individuals with STXBP1 that participated in the study were able to move independently.
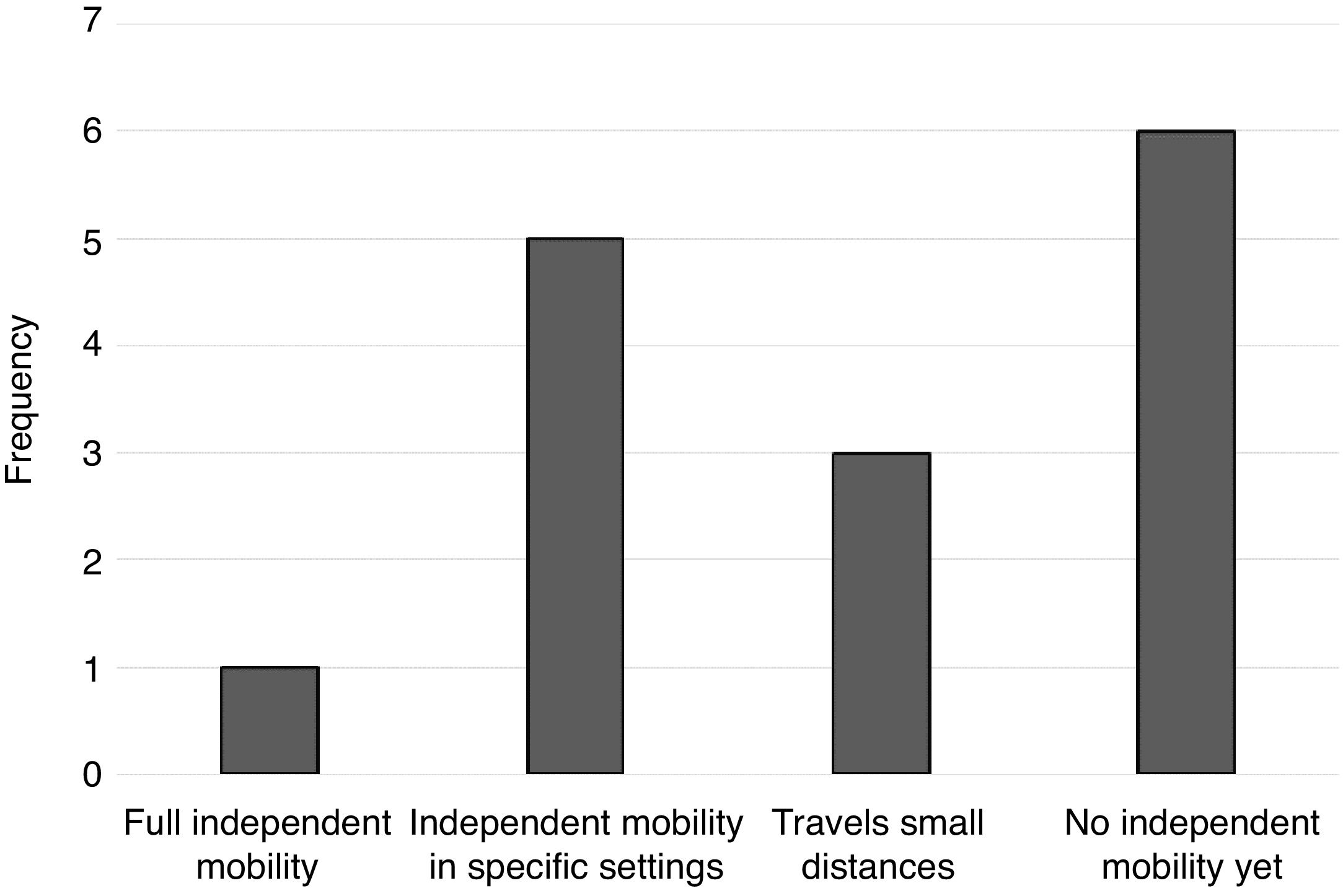
We found that 6% of participants exhibited fully independent mobility. Another 33% could move independently or with assistance in specific settings, such as the home or the school, and 20% could travel small distances independently, for instance, crawl to reach a toy. Lastly, 40% of participants could not move independently at all.
Other motor characteristicsIn addition to psychomotor retardation, most individuals with STXBP1 syndrome exhibit other motor characteristics that hinder coordination and voluntary movement. Fig. 3 shows the frequency of participants that exhibited rigidity, dyskinesia, ataxia and hypotonia. The cumulative sum of frequencies is greater than 15 because some individuals exhibited more than 1 of these features.
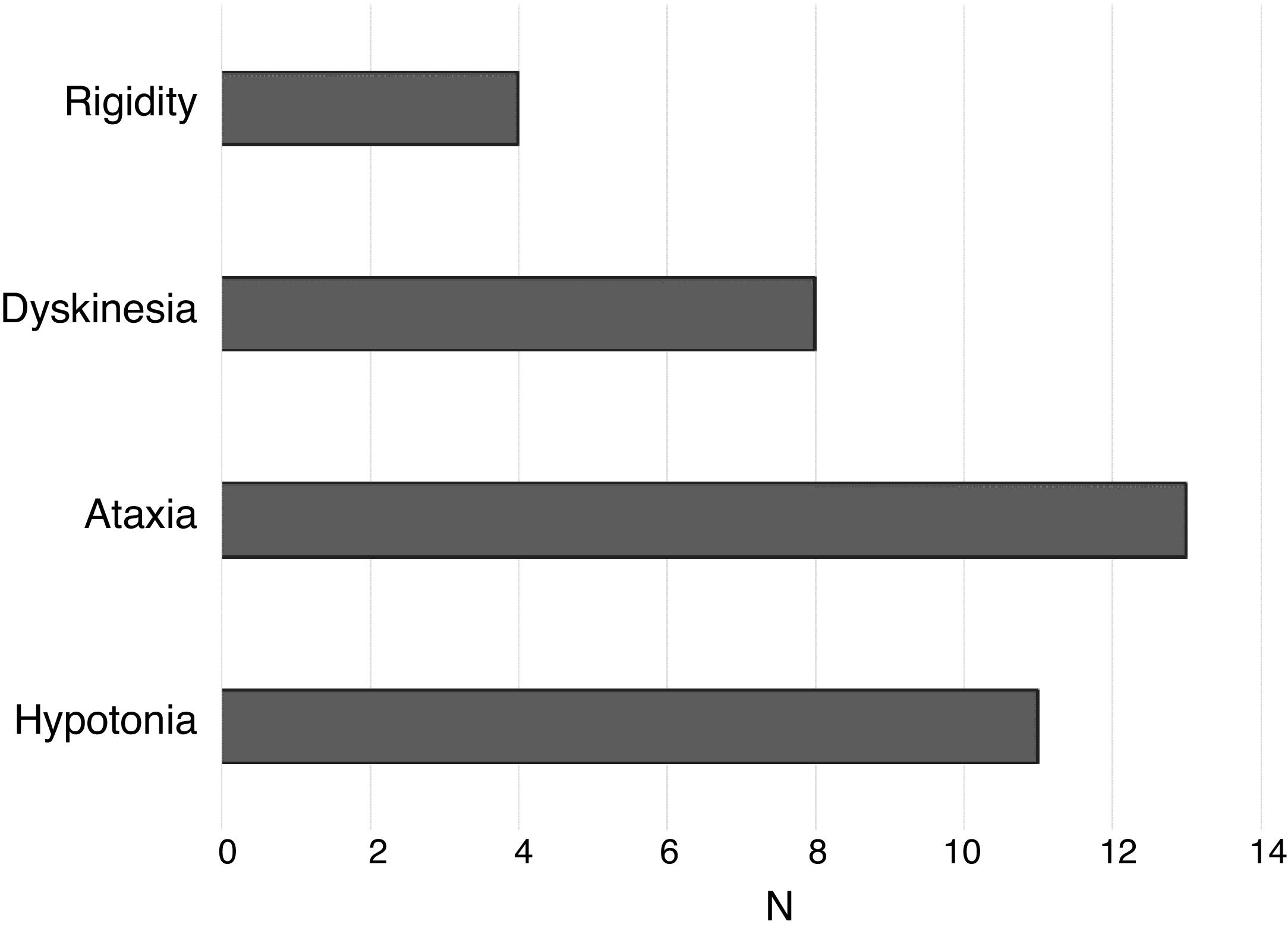
As can be seen in Fig. 3, the most frequent motor features were ataxia and hypotonia. Dyskinesia was also frequent, and rigidity was found in a lower proportion of participants.
Development of communication and social skillsIn terms of communication, most participants (67%) were at the pre-intentional stage. They showed little communicative intent so that individuals in their environment needed to interpret their intentions and wishes through observation. One participant had an additional diagnosis of ASD. The rest of participants exhibited communicative intent, and were able to use vocalizations, gestures or the gaze (13%), isolated words (13%) or full sentences (7%) to communicate.
When it came to the use of augmentative and alternative communication (AAC) support, at least 46% used some AAC system. Of those that did, 22% used signs, 45% pictograms and 33% communicators. Of the 11 participants that had not yet developed verbal language or vocalization and had only exhibited a limited communicative intent at the time of the study, 7 (63%) did not use any AAC system.
When it came to relationships with others, most participants in the study recognised and responded differently to individuals they knew. They enjoyed simple interactive games and asked to continue playing through different means. Half of the sample exhibited an interest in their peers, and 46% attempted interactions with their peers.
As for social relationships, we ought to mention that 1 of the participants has received a diagnosis of ASD and highlight the association of STXBP1 syndrome with ASD already mentioned in the introduction. Motor and cognitive impairments can sometimes mask specific problems in social relationships, so it is important to make a thorough evaluation of this dimension.
The diagnostic processThe 15 families that completed this phase of the study had received diagnosis within the past 5 years. Seven had received it within the past 6 months and 11 between 1 and 5 years before.
All participants developed the initial symptoms at an early age. The mean age of onset was 3 months, and the range a few days post birth to 13 months.
In most cases (10/15), the initial symptoms involved epileptic seizures, spasms or abnormal movements that warned of the possible presence of a disorder. However, despite the usual description of STXBP1 syndrome as an epileptic encephalopathy, in some cases the initial symptoms were not related to seizures, convulsions or spasms, but involved motor difficulties, psychomotor retardation, neurologic signs or uncontrollable crying. Far from being rare, in our sample 33% of the families reported onset with this type of features. Table 3 summarises the initial warning signs detected by the families.
Symptoms at onset of STXBP1 syndrome.
| Early symptoms | N.º |
|---|---|
| Epileptic seizures | 8 |
| Spasticity and spasms | 1 |
| Abnormal movements that did not disappear | 1 |
| Hypotonia | 2 |
| Stagnation of motor development (no standing, no sitting up, no crawling, no walking) | 2 |
| Uncontrollable crying | 1 |
| Hyperreflexia after birth | 1 |
Families received the definitive diagnosis from a paediatric neurologist in 57% of cases, a neurologist in 14% of cases and a geneticist in the remaining 29% of cases. Table 4 summarises the methods used to establish the diagnosis.
Although, as noted above, the onset occurred at an early age in all cases, there was a significant delay in the definitive diagnosis. This delay in diagnosis ranged from 7 months to 14 years.
When we took into account the initial symptoms that led to suspicion of a developmental problem, we found that the delay in diagnosis was much longer in cases in which the onset did not involve epileptic seizures, spasms or abnormal movements. Fig. 4 compares the mean delay in diagnosis in participants that had onset with epileptic seizures versus participants that did not.
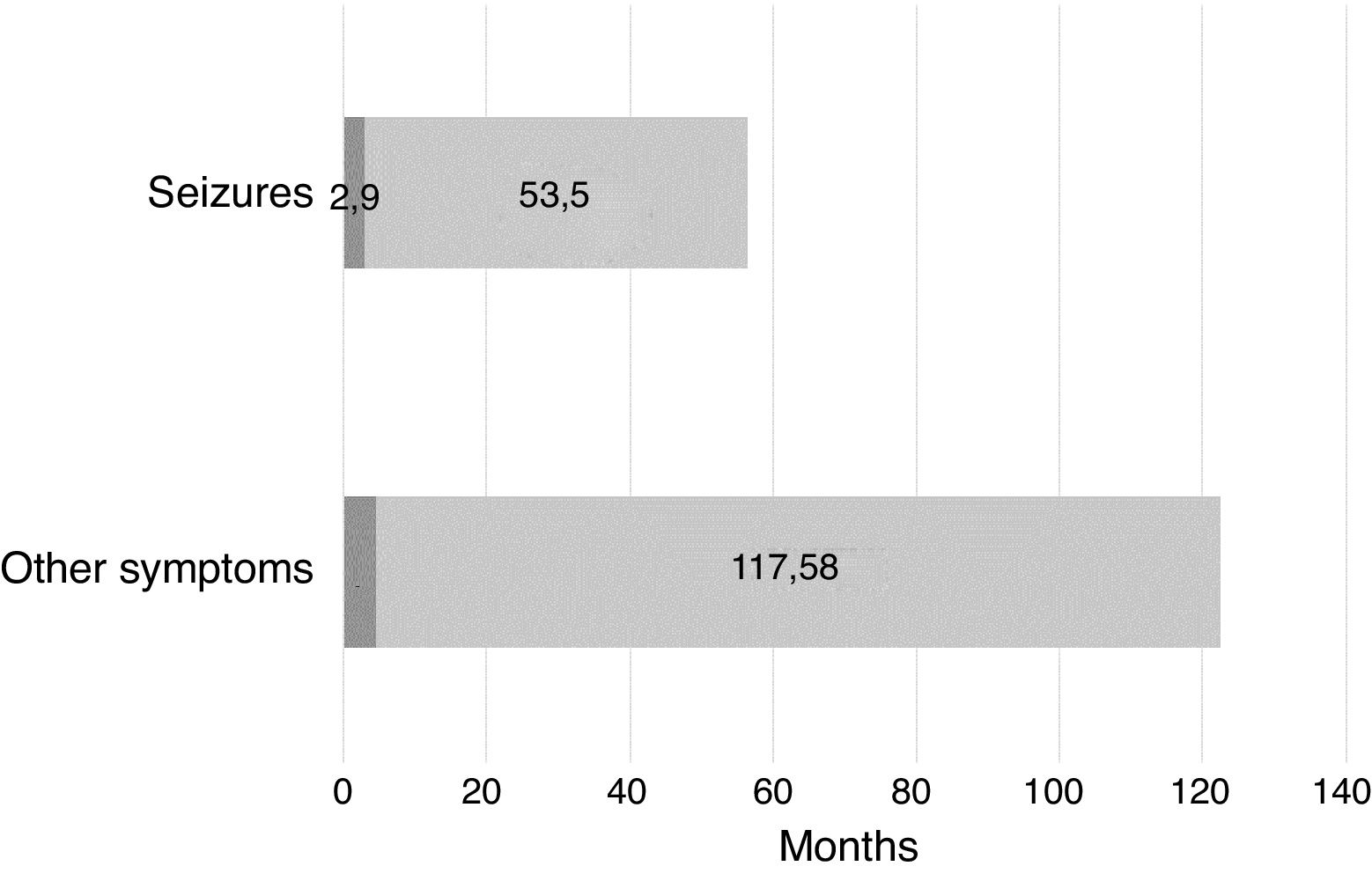
The subjective perception of families regarding the diagnostic process was consistent with the objective data, and evinced 2 clear needs: (1) a faster diagnosis to avoid unnecessary treatments, tests and evaluations and to develop a treatment plan as early as possible, and (2) more information on the disease to improve the understanding of what is happening and to be able to develop support networks.
DiscussionSTXBP1 syndrome manifests early in life and has a significant impact on neurodevelopment. Early diagnosis is essential to provide the necessary support, avoid unnecessary tests and counsel families on how to manage the necessary resources. To promote the dissemination of information on this syndrome and its early detection, we have gathered information from 17 families of affected individuals in Spain on the characteristics of patients with STXBP1 syndrome and the diagnostic process. The methods used in the study pose limitations, as we only have data from 17 families and we obtained the information directly from the families without comparing it to medical records. However, taking into account the low incidence of this syndrome and that these patients received the diagnosis relatively recently, we believe that the collected information may provide relevant data and a starting point for further research.
We now proceed to summarise the needs identified in the study in relation to the diagnostic process.
First, the number of cases of STXBP1 syndrome detected in Spain to date is much lower than expected based on currently available prevalence data. Taking into account the distribution of the population in Spain and the provinces where the identified affected individuals reside, it is evident that detection is associated with the accessibility of necessary tests or diagnostic services, and not to the actual incidence of the syndrome. The unequal geographical distribution of diagnosed cases suggests significant underdiagnosis in regions that do not have specialised services or facilities equipped to perform the necessary genetic tests. On the other hand, there is a growing knowledge of the phenotypic expression of changes in the STXBP1 gene, such as the occurrence of cases of mutations that never manifest with epileptic features.9 Thus, we expect that as more information becomes available and diagnostic tests more accessible, the number of cases identified in Spain will increase substantially. Although at present STXBP1 syndrome is considered a very rare disease, the emerging evidence suggests that these mutations could be involved in many cases of intellectual disability, movement disorders and epileptic encephalopathies of unknown cause.
At present, the delay in diagnosis is very high. Diagnosis is most delayed in cases presenting without epileptic seizures, spasms or abnormal movements serving as early warning signs of a disorder. Developmental stagnation and motor abnormalities could be the initial manifestations to prompt an early assessment of symptoms that would in turn guide the clinician to order the necessary genetic tests.
Most individuals with STXBP1 syndrome have experienced or will develop seizures at some point in life. Further research on the control of epilepsy is essential, on one hand to achieve control of seizures in refractory cases, and, on the other, to reduce intake of medication to the minimum possible. Given that in some cases epilepsy recurs after a long period of remission, research on the biological and behavioural markers that may provide information on the clinical course of epilepsy is also essential. This would help avoid the use of unnecessary medication or, on the other side of the spectrum, maintain treatment when there is risk of recurrence.
The characteristics of individuals with STXBP1 syndrome in our sample evince the substantial variation in the need for support in different areas. Further research is necessary in relation to communication and language development. Although previous studies have reported that 17% of individuals with STXBP1 syndrome also have ASD, only 1 person in our sample had this additional diagnosis. However, most affected individuals in our sample exhibited a limited communicative intent, and people in their environment needed to interpret their intentions and wishes. Although this aspect requires in-depth research, it is not clear that this lack of intent is due solely to cognitive or motor difficulties. On the contrary, these difficulties may mask specific disturbances in communication requiring further exploration. Consequently, it is essential that this dimension is investigated and to develop intervention strategies and information and technology systems to safeguard the right to communication of individuals with STXBP1 syndrome.
There is also little evidence on the efficacy of different interventions aimed at promoting motor development in affected individuals.
Improvements in the detection and diagnosis of STXBP1 syndrome and research on molecular, pharmacological and behavioural therapies in its management are essential to improve the quality of life of affected individuals and their families.
Conflicts of interestThe authors have no conflicts of interest to declare.
We thank the families in the STXBP1 Association for their invaluable collaboration in this study. Information on this association and on how to contact it can be found at: https://stxbp1.es/
Please cite this article as: Murillo E. Características de las personas con el síndrome STXBP1en España: implicaciones para el diagnóstico. An Pediatr (Barc). 2020;92:71–78.
