




Adrenocortical (AC) tumours are extremely rare in children, and there is little evidence on the management of paediatric cases. We present the only 2 cases of AC carcinoma managed in our hospital.
Case 1
Boy aged 11 years with an unremarkable history in whom the diagnostic evaluation of epileptic seizures detected a mass in the right adrenal gland. The patient also presented with hypertension and increases in body hair growth, weight, height and acne associated with elevation of androgens (testosterone, 5.9 ng/mL; dehydroepiandrosterone sulphate [DHEA-S], 5770 ng/mL; androstenedione, 940 ng/dL), estradiol (21 pg/mL), 17-OH progesterone (298 ng/dL) and prolactin (24.2 ng/mL). The patient underwent partial resection of the solid mass, which measured 80 × 80 × 97 mm and showed no sign of infiltration, with enlargement of adjacent lymph nodes to 10 and 14 mm, features compatible with AC carcinoma. The initial staging found no spread. Hormone levels normalised following surgery, but the positron emission tomography/computed tomography (PET/CT) scan was positive, leading to initiation of radiation therapy and mitotane. The patient had developed lymph node metastasis at 5 months of treatment, leading to second-line chemotherapy with mitotane, cisplatin, etoposide and doxorubicin, which achieved partial remission, and consolidation therapy consisting of allogeneic haematopoietic stem cell transplantation and NK cell infusion. The patient died 1 year after diagnosis due to complications of transplantation.
Case 2
Girl aged 14 years with an unremarkable history that presented with dysmenorrhoea, acne and hirsutism. The endocrine evaluation evinced elevation of testosterone (5.34 ng/mL) and DHEA-S (44 000 ng/mL). The MRI at diagnosis evinced a large retroperitoneal mass (Fig. 1) with a negative PET/TC scan. The patient underwent complete resection of the tumour, which measured 16 × 15 × 4 cm, weighed 850 g and had an intact capsule and necrotic and bleeding areas, features compatible with AC tumour of unknown malignant potential (p53 labelling index <50% positive cells; negative results for CD10, epithelial membrane antigen [EMA] and keratin clone AE1/AE3; Ki67 labelling index 10%, <15 mitoses per 20 HPF and absence of atypical mitotic figures). Hormone levels normalised and treatment with mitotane was initiated after surgery, and the patient is in complete remission at 1 year of treatment.
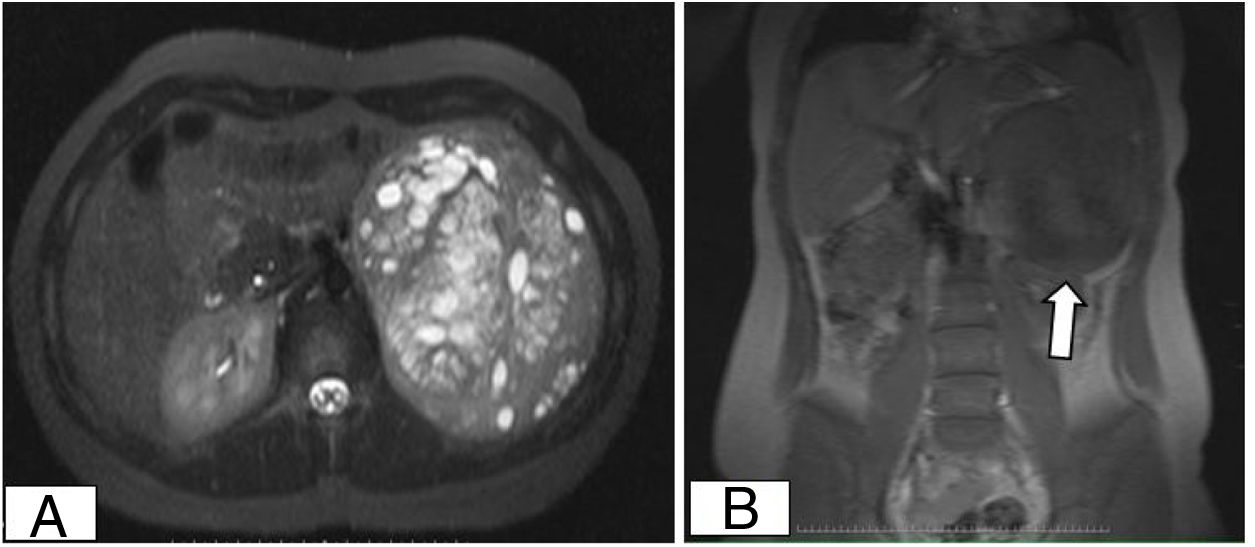
Abdominal magnetic resonance image showing a large retroperitoneal mass with clearly defined borders. (A) Axial view showing uptake of contrast in most of the mass with areas of necrosis and a small area with bleeding. (B) Coronal view showing contact of the mass with the spleen and the left kidney.
Adrenocortical tumours amount to 0.2% of malignant tumours in children. They are more prevalent in women and have a bimodal age distribution, with incidence peaking in the first 5 years and the fifth decade of life.1 However, they may develop during adolescence, as occurred in the 2 cases presented here.
The pathogenesis of AC tumours is partially known, and their development, in isolation or associated with other diseases like Li-Fraumeni syndrome, is associated with changes in the p53 tumour suppressor gene.2 Ninety percent of cases have onset with isolated virilization (case 2), followed by hypersecretion of mineralocorticoids or glucocorticoids (case 1), which carries a poorer prognosis. Up to 10% of AC tumours are non-functioning, a presentation that is more frequent at advanced ages.1
The differential diagnosis of adenoma and carcinoma is challenging from both a radiological and a histological perspective, and the Wienecke score is applied for the purpose3 (Table 1). As regards prognostic factors, in addition to the stage of the tumour and extent of surgical resection, Picard et al. described poorer outcomes in patients with AC tumours with a Ki67 index greater than 15% and highlighted the predictive value of this parameter combined with the Wieneke criteria for the purpose of establishing the indication of adjuvant therapy.4 The 5-year overall survival is less than 30% in patients with carcinoma.5
Wieneke criteria.
| 1. Tumour weight > 400 g |
| 2. Tumour size > 10.5 cm |
| 3. Extension into adjacent organs. |
| 4. Invasion into the vena cava |
| 5. Venous invasion |
| 6. Capsular invasion |
| 7. Presence of tumour necrosis |
| 8. Mitotic count > 15 per 20 high power fields |
| 9. Presence of atypical mitotic figures |
| Prognostic groups: |
| • ≤ 2 criteria = benign |
| • 3 criteria = uncertain malignant potential |
| • ≥ 4 criteria = malignant |
Guidelines have yet to be established to standardise treatment due to the low frequency of this disease in the paediatric age group. The cornerstone of treatment is surgical resection, and complete resection is the strongest predictor of a favourable outcome. Chemotherapy is indicated in patients with disseminated disease or meeting histological criteria of malignancy, and the most frequently used drug is mitotane. Radiation therapy is used in case of symptomatic local recurrence, incomplete resection or bone or brain metastases. At present, there are no open trials for treatment of adrenocortical tumours in Spain.
In both cases presented here, testing was performed to screen for p53 variants, and a change was only detected in case 1. This patient received radiation therapy and chemotherapy post surgery in adherence with the ARAR0332 protocol recommended by the Children’s Oncology Group,6 as the patient had a stage III tumour, followed by salvage systemic therapy due to metastatic dissemination. The second patient, despite complete resection of the tumour and normalization of hormone levels after surgery, received adjuvant therapy with mitotane as the mass was a functioning tumour of uncertain malignant potential (it met 3 of the Wieneke criteria: tumour weight >400 g, tumour size >10.5 cm and presence of necrosis).
Given the low prevalence and potential severity of AC tumours, it would be beneficial to facilitate performance of multidisciplinary evaluation in referral hospitals with experience in the surgical management and adjuvant treatment of this disease.
Please cite this article as: Sánchez Sierra N, Buendía López S, Andión Catalán M, Camarena Pavón N, Lassaletta Atienza A. Tumores corticosuprarrenales: presentación de 2 casos. An Pediatr (Barc). 2021;95:267–268.
Previous presentation: the study was presented at the XII National Congress of the Sociedad Española de Hematología y Oncología Pediátricas, May 30–June 1, 2019; Jerez de la Frontera, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals