

Pediatric acute liver failure (ALF) due to inherited metabolic diseases (IMD) is a rare life-threatening condition with a poor prognosis. Early intervention may be lifesaving.
ObjectiveTo describe clinical presentation, investigation and outcomes of ALF related to IMD in young children.
Material and methodsRetrospective review of the medical records of children aged up to 24 months, admitted to a tertiary pediatric and neonatal Intensive Care Unit during a 27-year period, fulfilling the ALF criteria, with documented metabolic etiology.
ResultsFrom 34 ALF cases, 18 were related to IMD: galactosemia (4), mitochondrial DNA depletion syndrome (MDS) (3), ornithine transcarbamilase deficiency (3), congenital defects of glycosylation (2), tyrosinemia type 1 (2), long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (1), hereditary fructose intolerance (1), classic methylmalonic aciduria (1) and citrulinemia type 1 (1). The median age was 1.3 months. At least one previous suggestive sign/symptom of IMD (vomiting, failure to thrive, hypotonia or developmental delay) was observed in 67% of the cases. The most common physical signs at admission included: hepatomegaly (72%), jaundice (67%) and encephalopathy (44%). The peak laboratorial findings were: mean international normalizad ratio 4.5, median lactate 5mmol/L, mean bilirubin 201μmol/L, median alanine aminotransferase (ALT) 137UI/L and median ammonia 177μmol/L. One patient was submitted to liver transplant in ALF context (MSD). The mortality rate was 44%.
DiscussionThe identification of IMD as a frequent cause of ALF allowed specific therapeutic measures and adequate family counseling. Particular clinical features and moderated ALT and bilirubin levels can lead to its suspicion.
La insuficiencia hepática aguda (IHA) secundaria a enfermedades metabólicas hereditarias (EMH) es una enfermedad grave infrecuente de mal pronóstico. La intervención temprana puede salvar vidas.
ObjetivoDescribir la presentación clínica, la investigación y la evolución de IHA asociada a EMH en niños pequeños.
Material y métodosEstudio retrospectivo de las historias clínicas de niños con edad de hasta 24 meses ingresados en una unidad terciaria de cuidados intensivos neonatales y pediátricos, durante un período de 27 años, que cumplían los criterios diagnósticos de IHA y con etiología metabólica documentada.
ResultadosDe los 34 casos de ALF, 18 se asociaban a EMH: galactosemia (4), síndrome de depleción del ADN mitocondrial (SDM) (3), deficiencia de ornitina transcarbamilasa (3), defectos congénitos de la glucosilación (2), tirosinemia tipo 1 (2), deficiencia de 3-hidroxi-acil-CoA deshidrogenasa de cadena larga (1), intolerancia hereditaria a la fructosa (1), aciduria metilmalónica clásica (1) y citrulinemia tipo 1 (1). La edad mediana fue de 1,3 meses. En el 67% de los casos se había observado al menos un síntoma o signo indicativo de EMH con anterioridad (vómitos, fallo de medro, hipotonía o retraso del desarrollo). Los signos físicos más frecuentes en el momento del ingreso fueron: hepatomegalia (72%), ictericia (67%) y encefalopatía (44%). Los niveles analíticos pico fueron: razón internacional normalizada media, 4,5; lactato mediano, 5mmol/L; bilirrubina media, 201μmol/L; alanina aminotransferasa (ALT) mediana, 137 UI/L; y amonio mediano, 177μmol/L. Un paciente fue remitido para trasplante de hígado en contexto de IHA (MSD). La tasa de mortalidad fue del 44%.
DiscusiónLa identificación de EMH como causa frecuente de IHA permitió el uso de medidas terapéuticas específicas y un asesoramiento familiar apropiado. Sus manifestaciones clínicas particulares y niveles moderados de ALT y bilirrubina pueden llevar a sospechar esta condición.
Pediatric acute liver failure (ALF) is a rare life-threatening condition that may progress quickly, with severe impairment of hepatic function. It can be recognized by hepatocellular necrosis and refractory coagulopathy with or without encephalopathy. The etiology of ALF varies according to the age of the patient and geographic areas. In neonates and infants, viral infections (enteroviruses, herpes simplex and others), neonatal hemochromatosis, inherited metabolic diseases (IMD) and hemophagocytic lymphohistiocytosis are the main causes. In older children, the most common causes are drug-induced ALF, autoimmune hepatitis and viral infections, being hepatitis A virus the most frequent in developing countries.1 Inherited metabolic diseases deserve special attention in differential diagnosis of pediatric ALF, particularly in infants and young children, in whom they constitute 13–43% of all cases.2 In the first 3 years of life, the commonest IMD related to ALF are: galactosemia; tyrosinemia type 1; mitochondrial cytopathy; fatty acid oxidation defect; hereditary fructose intolerance; urea cycle defect and congenital disorders of glycosylation.2 In this particular age group, it is fundamental to conduct an exhaustive, structured and expeditious diagnostic work-up to minimize indeterminate cases that could be explained by an unrecognized IMD.
The prognosis of IMD related ALF is generally poor and depends on the specific diagnosis.3 Early recognition of these etiologies is essential, since some require disease-specific treatment or may constitute a contraindication to liver transplantation (LT), as in cases with multisystemic involvement.1,2,4 LT can improve the outcome in IMD cases who fail to respond to conservative management.2,5
To improve IMD diagnosis and intervention in ALF cases, a better knowledge of their clinical presentation and biochemical markers is fundamental. The aim of this study was to characterize the clinical presentation, laboratory profile and outcome of young children admitted to a pediatric and neonatal intensive care unit (ICU) with ALF related to IMD, during a 27-year period.
MethodsThis is a descriptive study conducted in the pediatric and neonatal ICU of a tertiary university hospital, the national referral center for pediatric liver transplantation. Medical records of all children aged up to 24 months admitted to ICU during a 27-year period, (between January 1989 and December 2015), fulfilling ALF criteria and with a documented IMD were reviewed.
Acute liver failure was defined according to Pediatric Acute Liver Failure Study Group6 by the following criteria: (1) children with no known evidence of chronic liver disease; (2) biochemical evidence of acute liver injury, and (3) presence of hepatic-based coagulopathy, with international normalized ratio (INR)≥1.5 not corrected by vitamin K in the presence of hepatic encephalopathy (HE) or INR≥2 regardless of the presence of HE. Encephalopathy was evaluated by electroencephalogram and/or by clinical signs, classified in four grades: grade 1 – changes in behavior, minimal change in level of consciousness and altered sleep; grade 2 – drowsiness, confusion, inappropriate behavior, disorientation, mood swings; grade 3 – somnolent but arousable, gross disorientation, bizarre behavior, muscular rigidity, clonus and hyperreflexia and grade 4 – coma and decerebration.1,7
Demographic data, family background, medical history and clinical signs and symptoms at admission were analyzed. Peak levels of white blood cell (WBC) count, INR, lactate, ammonia, total bilirubin and alanine aminotransferase (ALT), as well as initial glycemia and minimum levels of albumin were recorded. Baseline metabolic work-up included: acyl-carnitine profile, plasma and urinary amino acids and urinary organic acids profiles, plasma carbohydrate-deficient transferrin, urinary reducing substances and ketones. Other laboratory and metabolic investigations were carried out in a case by case approach. DNA extraction was performed for specific molecular/genetic studies. Other information collected included results of molecular tests, timing of the confirmation of etiological diagnosis (in vivo or post-mortem), decision on liver transplantation, time to ICU discharge, mortality to discharge and overall mortality.
Statistical Package for the Social Science® version 20 was used for statistical analysis. Quantitative variables were evaluated by measures of central tendency and of dispersion and qualitative variables by determination of frequencies.
ResultsDuring the study period, from a total of 34 children aged up to 24 months admitted at ICU with ALF, in 18 cases an IMD was confirmed. The median age was 1.3 months (Q1:0.2; Q3:5.5), 8/18 were newborns and 8/18 were boys.
The etiological IMDs identified and the results of genetic investigation are depicted in Table 1. In the case of hereditary fructose intolerance, the diagnosis was established by enzymatic study in a liver biopsy, which showed reduced capacity of aldolase to split fructose-1-P and a ratio of fructose-1,6-bisphosphate/fructose-1-P of 38 (N≈1). The youngest patients were diagnosed during the neonatal period with galactosemia (3), mitochondrial DNA depletion syndrome (MDS) (2), ornithine transcarbamilase (OTC) deficiency (1), classic methylmalonic aciduria (cMMA) (1) and citrulinemia type 1 (1).
ALF etiologies and genetic investigations.
| IMD | n | Genetic investigation |
|---|---|---|
| Galactosemia | 4 | GALT gene: Q188R (homozygous) (2) GALE gene: p.Val94Mel c.280G>A (homozygous) (2) |
| MDS | 3 | DGUOK gene: c.677A>G (homozygous) (2) and c.749T>C (homozygous) (1) |
| OTC deficiency | 3 | OTC gene: Tyr143 stop exon 5 (1); R92Q CGA>CAA codon 92 (1) and R23X CGA>TGA codon 23, exon 1 (homozygous) (1) |
| CDG | 2 | CCDC115 gene: p.leuc31Ser c.92T>C (1) |
| Tyrosinemia type 1 | 2 | FAH gene: IVS6-1(G>T)/IVS7+1(G>A) (1) and IVS6-1(G>T) (homozygous) (1) |
| LCHAD deficiency | 1 | HADHA gene: G1528C (homozygous) |
| Hered. fructose intolerance | 1 | – |
| cMMA | 1 | MUT gene: c.1172delA (p.D391Vfs*9) (homozygous) |
| Citrulinemia type 1 | 1 | ASS1 gene: c.970G>A (p.G324S) (homozygous) |
ALF: acute liver failure; CDG: congenital disorder of glycosylation; IMD: inherited metabolic diseases; LCHAD: long-chain 3-hydroxyacyl-CoA dehydrogenase; MDS: mitochondrial DNA depletion syndrome; cMMA: classic methylmalonic aciduria; OTC: ornithine transcarbamylase.
Information about consanguinity was available in 13/18, being positive in 3. Family history was unremarkable in all but two cases: one of a patient with MDS, whose brother died in another hospital with the diagnosis of neonatal hemochromatosis, later proved to also be a MDS due to deoxyguanosine kinase (DGUOK) deficiency; and a case of a newborn, sister of an elder patient already included in this study, who died with ALF and whose post-mortem investigation allowed to make the diagnosis of galactosemia due to galactose epimerase (GALE) deficiency in both siblings.
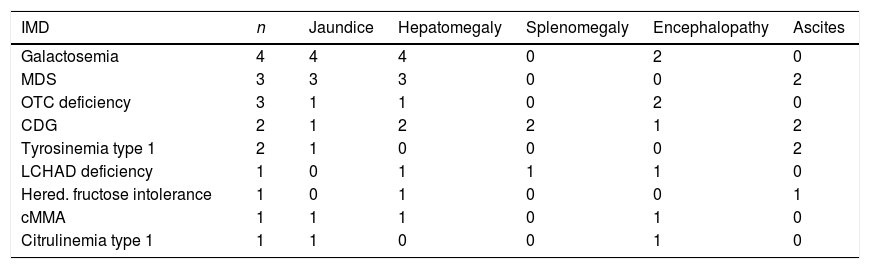
Retrospectively, 67% (12/18) of the cases had at least one of the following signs/symptoms: vomiting; failure to thrive; hypotonia and developmental delay. The most common physical signs at admission included: hepatomegaly (13/18; 72%); jaundice (12/18; 67%); encephalopathy (8/18; 44%); ascites (7/18; 39%) and splenomegaly (3/18; 17%). 5/8 patients had severe encephalopathy (grades 3–4). The physical signs at admission, according to the underlying IMD, are described in Table 2.
Presenting physical signs of ALF according to etiology.
| IMD | n | Jaundice | Hepatomegaly | Splenomegaly | Encephalopathy | Ascites |
|---|---|---|---|---|---|---|
| Galactosemia | 4 | 4 | 4 | 0 | 2 | 0 |
| MDS | 3 | 3 | 3 | 0 | 0 | 2 |
| OTC deficiency | 3 | 1 | 1 | 0 | 2 | 0 |
| CDG | 2 | 1 | 2 | 2 | 1 | 2 |
| Tyrosinemia type 1 | 2 | 1 | 0 | 0 | 0 | 2 |
| LCHAD deficiency | 1 | 0 | 1 | 1 | 1 | 0 |
| Hered. fructose intolerance | 1 | 0 | 1 | 0 | 0 | 1 |
| cMMA | 1 | 1 | 1 | 0 | 1 | 0 |
| Citrulinemia type 1 | 1 | 1 | 0 | 0 | 1 | 0 |
ALF: acute liver failure; CDG: congenital disorder of glycosylation; IMD: inherited metabolic diseases; LCHAD: long-chain 3-hydroxyacyl-CoA dehydrogenase; MDS: mitochondrial DNA depletion syndrome; MMA: classic methylmalonic aciduria; OTC: ornithine transcarbamylase.
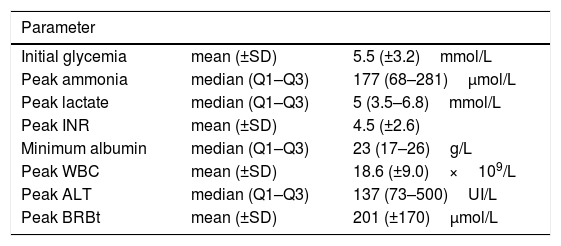
Data concerning mean or median peak levels of WBC count, INR, lactate, ammonia, total bilirubin, ALT, initial glycemia and minimum levels of albumin are shown in Table 3. Etiological diagnosis was confirmed by further investigations requested on a child-to-child basis, except in 5 cases in which the diagnostics were done in the post-mortem period [2 MDS, 1 long chain Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD), 1 congenital defect of glycosylation (CDG) and 1 galactosemia due to GALE deficiency].
Laboratorial parameters according to etiology.
| Parameter | ||
|---|---|---|
| Initial glycemia | mean (±SD) | 5.5 (±3.2)mmol/L |
| Peak ammonia | median (Q1–Q3) | 177 (68–281)μmol/L |
| Peak lactate | median (Q1–Q3) | 5 (3.5–6.8)mmol/L |
| Peak INR | mean (±SD) | 4.5 (±2.6) |
| Minimum albumin | median (Q1–Q3) | 23 (17–26)g/L |
| Peak WBC | mean (±SD) | 18.6 (±9.0)×109/L |
| Peak ALT | median (Q1–Q3) | 137 (73–500)UI/L |
| Peak BRBt | mean (±SD) | 201 (±170)μmol/L |
ALT: alanina aminotransferase; BRBt: total bilirubin; INR: international normalized ratio; SD: standard deviation; WBC: white blood cells.
The retrospective analysis of physical and laboratorial features allowed the identification of some particular characteristics in patients with the same IMD. Both patients with tyrosinemia type 1 presented renal tubulopathy. The 4 cases of urea cycle disorders (OTC deficiency and citrulinemia type 1) presented high levels of ammonia when compared with other etiologies: median 333μmol/L (Q1:118; Q3:1703) vs 172.5μmol/L (Q1:61.8; Q3:236.5), respectively. In the galactosemia due to GALE deficiency, hypotonia, bilateral hip dysplasia and short stature were features common to both cases. Short stature, some facial dysmorfisms and hepato-splenomegaly were present in both cases of CDG. The 3 patients with MDS due to DGUOK deficiency revealed a positive history of consanguinity, hypotonia, nystagmus, hypoglycemia and high levels of ferritin [789;1931]ng/mL (normal range: 9–120) and tyrosine [68;543]μmol/L (normal range: 28–96). The patient with LCHAD presented with cardiogenic shock.
Liver transplant was performed in ALF context in one patient with MDS. It was decided by a multidisciplinary team at the age of 4 months due to recurrent liver failure, before the molecular confirmation of DGUOK deficiency, despite neurologic dysfunction and rotatory nystagmus. However, he died at 4 years of age due to an intercurrence with metabolic acidosis, hyperlactacidemia and cardiorespiratory arrest. Another patient with tyrosinemia was submitted to LT later on, at the age of 7 years, due to unsatisfactory metabolic control, osteopenia, renal tubulopathy and hepatic nodules.
Analyzing the outcomes, the median time to ICU discharge was 7 days (Q1:3; Q3:21). The mortality until discharge and overall mortality were 39% (7/18) and 44% (8/18), respectively. Besides the MDS patient mentioned above, the diagnosis of the other 7 deceased patients were: MDS (2), LCHAD (1), CDG (1), galactosemia due to GALE deficiency (1), OTC deficiency (1) and cMMA (1). The major causes of death were due to progression to multiorgan failure or cardiogenic shock. In 3 of these cases (CDG, OTC deficiency and cMMA), the adjustment of therapeutic effort was decided in multidisciplinary consensus, based on the conditions described hereafter: (1) In the CDG case, despite optimized therapeutic measures including consideration for LT, the multisystemic involvement with unfavorable neurological evolution due to severe hypoxic ischemic lesions documented by imaging studies precluded this option; (2) In the case with OTC deficiency, the high ammonia (>800μmol/L) levels were difficult to control during the first 48hours, despite maximum therapeutic measures, including hemodiafiltration. Although ammonia levels decreased afterwards, no neurological improvement was achieved. The patient progressed to multiorgan failure, exacerbated by a Klebsiella oxytoca sepsis; (3) In the cMMA case, despite prompt providing of specific therapeutics and reduction of ammonia levels, the clinical evolution was unfavorable since admission with rapid progression to multiorgan failure.
DiscussionThe disorders that can lead to liver failure in the pediatric age group up to 24 months are quite specific and IMDs represent one of the most frequent etiologies, with a described frequency of 13–43%.2 In the present study, from a total of 34 children with ALF in this age group, 18 (53%) were diagnosed with an IMD. This high number of identified IMDs results from a progressive effort to carry out a more comprehensive diagnostic investigation, even in the post-mortem period. It can also be explained by the recognition of this tertiary hospital as the unique referral center in Portugal for LT and ALF since 1994 and 2008, respectively. Patients without a readily identifiable cause of ALF must be considered for further screening for metabolic causes of ALF, especially in this age group. Pediatric ALF etiology remains indeterminate in 18–47%,1 depending upon the exhaustiveness of work-up screening, and it can even be higher in younger children, approaching 54% in children under 3 years, as reported by Squires et al.6
Early recognition of IMDs is crucial, because some require immediate initiation of specific therapy or diets that can be life-saving, mainly in the case of galactosemia, hereditary fructose intolerance and hereditary tyrosinemia type 1,1 which can present during neonatal period with jaundice, hypoglycemia and sometimes with ALF.8 In this case series, the lower number of patients with galactosemia and hereditary tyrosinemia type 1 presenting with ALF, compared to King's College experience,9 may be attributed to the expanded Portuguese neonatal screening, available since 2005, which allows pre-symptomatic diagnosis of these diseases.
The scarce consanguinity information available (in only 13/18) is a limitation related to the retrospective nature of this study. According to Hegarty et al.,9 history of parental consanguinity can be present in 44% of IMD related ALF. Particularities of the familial history, such as recurrent abortions, sibling deaths and previously affected children, are other features that must always be explored, since they can raise the suspicion of IMDs, as found in the cases of MDS and galactosemia due to GALE deficiency.
The previous history of vomiting, failure to thrive and/or developmental delay, present in 67% of cases, is another strong pointer suggesting an IMD, as suggested by Alam et al.,2 and Brett et al.4 Encephalopathy was more frequent (44%) when compared to Hegarty's IMD series9 (19%), a fact that can be explained by the different ratio of each specific IMD in both samples. Other reason that could contribute to this result was the possible delay in referral to our center occurred prior to 2008, date of its inception as national referral center for ALF.10 Some clinical features can provide clues to a specific IMD, guiding the etiological investigation.11 The cardiac involvement can lead to LCHAD suspicion. Some dysmorphisms are known to be more frequent in CDG and in galactosemia due to GALE deficiency.11,12 The triad of hypotonia, nystagmus and hypoglycemia is typically present in MDS due to DGUOK deficiency cases. These patients can also have high levels of ferritin and tyrosine, leading to the differential diagnosis of neonatal haemochromatosis and tyrosinemia type 1.13 Other clues from laboratorial results are signs of tubulopathy, that may suggest tyrosinemia type 1, classic galactosemia or hereditary fructose intolerance and marked hyperammonemia which is a typical finding in urea cycle disorders.11
Regarding the laboratorial parameters, the slight to moderate elevation of ALT observed is consistent to levels found in other studies of IMD related ALF9,14 and inferior than in other etiologies, especially in viral infection.14 The moderate elevation of bilirubin levels was also similar to findings of other studies.9,14 In two small series4,15 that compared laboratory profile in IMD related ALF with other causes, peak levels of lactate and ammonia were similar between both groups and also identical to levels found in this study, suggesting that they may not be useful as isolated markers of IMD in ALF.
The recognition of IMD is crucial to optimize the management with targeted intervention, to evaluate the need for LT and to do adequate parental counseling in future pregnancies.16 Decisions on transplantation are extremely difficult, a fact that is corroborated by the lack of validated pediatric ALF prognostic models,17 especially for IMD etiology. LT may be contraindicated in cases in which ALF may resolve with specific medical and/or diet therapy, if the underlying disease involves other organs or systems or if there is a rapid progression of ALF with multiorgan failure or sepsis.5,18 Of the 18 patients included in this study only 1 was submitted to LT in ALF context, a patient with DGUOK deficiency that received a LT, despite the presence of neurologic dysfunction and rotatory nystagmus. However, it did not prevent disease progression, with neurologic and visual impairment, severe psychomotor delay and failure to thrive. In this hepato-cerebral mitochondrial DNA depletion syndrome, the contra-indication for LT is unclear, since there are also some descriptions in the literature of a significant proportion of patients who benefit from LT with good long-term survival and a stable neurological situation despite initial neurological abnormalities.19,20
The overall mortality rate of 44% was lower when compared to the 70% reported by Alam et al.,15 but higher than the 19% described by Hegarty.9 Sundaram et al.,14 reported a 24% of mortality without LT. However, the comparison of mortality rates is hampered by group heterogeneities concerning the age, the different etiologies of ALF and the small case series numbers. In fact, while Sundaram14 studied ALF in infants up until 3 months of age, in Hegarty's series9 children were aged up to 5 years old and the majority of diagnosis were galactosemia or tyrosinemia, to which a good prognosis is generally attributed. In our study, the high percentage of patients with multisystem involvement or with rapid progression to multiorgan failure could explain the mortality rate. In addition, 44% patients presented with encephalopathy, which is generally recognized as a poor prognostic marker in ALF.21
Over the last years, the implementation of expanded newborn screening programs in several countries, as well as the availability of new specific therapies for some diseases and advanced supportive devices have contributed to modify some IMD's natural history and progression.1,4 As shown in Couce series22 about the incidence of IMD in a neonatal unit, not necessary related to ALF, the investment in an early neonatal screening can allow the early diagnosis and treatment in a group of patients in a pre-symptomatic period, avoiding severe clinical complications and hospital admission in a number of cases.
The main limitations of this study are related to the small number of cases, the large period considered and the retrospective data collection, that could limit the information obtained. Moreover, only ALF cases admitted to ICU were included in this series, representing the most severe ones. This sample selection could create a bias.
This study points out the high frequency of IMD related ALF in this particular age group and the importance of an exhaustive diagnostic work-up to reach a correct diagnosis. The effort to have a post-mortem diagnosis allows for an adequate family counseling. Some clinical and laboratorial features, such as young age and moderated elevation of ALT and bilirubin peak levels, may raise the suspicion of IMD related ALF. The early diagnosis of this etiology can lead to a prompt therapeutic decision, contributing to achieving a better outcome. Future directions must strive to optimize therapeutic decisions regarding LT, which may be achieved by large and homogeneous multicentric studies.
Conflict of interestThe authors declare that they have no conflict of interest.
Please cite this article as: Dias Costa F, Moinho R, Ferreira S, Garcia P, Diogo L, Gonçalves I, et al. Insuficiencia hepática aguda asociada a enfermedades metabólicas hereditarias en niños pequeños. An Pediatr (Barc). 2018;88:69–74.
