



Achondroplasia requieres multidisciplinary follow-up, with the aim of preventing and managing complications, improving the quality of life of people who suffer from it and favoring their independence and social inclusion. This review is justified by the multiple publications generated in recent years that have carried out a change in its management. Different guidelines and recommendations have been developed, among which the one made by the American Academy of Pediatrics in 2005 recently updated (2020), the Japanese guide (2020), the first European Consensus (2021) and the International Consensus on the diagnosis, approach multidisciplinary approach and management of individuals with achondroplasia throughout life (2021). However, and despite these recommendations, there is currently a great worldwide variability in the management of people with achondroplasia, with medical, functional and psychosocial consequences in patients and their families. Therefore, it is essential to integrate these recommendations into daily clinical practice, taking into account the particular situation of each health system.
La acondroplasia requiere un seguimiento multidisciplinario, con el objetivo de prevenir y manejar las complicaciones, mejorar la calidad de vida de las personas que la padecen y favorecer su independencia e inclusión social. Esta revisión se justifica por las múltiples publicaciones generadas en los últimos años que han llevado a cabo un cambio en su gestión. Se han desarrollado diferentes guías y recomendaciones, entre las que destacan la realizada por la Academia Americana de Pediatría en 2005 recientemente actualizada (2020), la guía japonesa (2020), el primer Consenso Europeo (2021) y el Consenso Internacional sobre el diagnóstico, abordaje, enfoque multidisciplinario y manejo de individuos con acondroplasia a lo largo de la vida (2021). Sin embargo, y a pesar de estas recomendaciones, actualmente existe una gran variabilidad a nivel mundial en el manejo de las personas con acondroplasia, con consecuencias médicas, funcionales y psicosociales en los pacientes y sus familias. Por ello, es fundamental integrar estas recomendaciones en la práctica clínica diaria, teniendo en cuenta la situación particular de cada sistema sanitario.
Achondroplasia is the most frequent skeletal dysplasia associated with disproportionate short statute. Its incidence is estimated at 1 case per 10 000–30 000 live births, with no differences based on sex or race. A total of 360 000 individuals worldwide have this rare disease.1 The global prevalence is of 4.73 cases per 100 000 individuals, with a prevalence of 3.62 cases per 100 000 individuals in Europe.2 This form of dysplasia is caused by a heterozygous pathogenic variant in the FGFR3 gene in chromosome 4p16.3, which encodes fibroblast growth factor receptor 3 (FGFR3), resulting in gain of function of the receptor3 through the activation of the mitogen activated protein kinase (MAPK) signalling pathway, thereby inhibiting the proliferation and differentiation of chondrocytes in the growth plate.4 Other pathways have been found to be implicated, such as the STAT, Wnt/β-catenin, PI3K/AKT and PLCγ.5
Achondroplasia causes a constellation of skeletal comorbidities resulting from abnormal endochondral ossification (long bones and axial skeleton). It produces a characteristic phenotype with disproportionate short stature due to a rhizomelic shortening of the extremities and a sitting height that approximates the reference height.6 Affected individuals exhibit macrocephaly, a prominent forehead, hypoplasia of the midface and hands with a three-pronged appearance. The average expected adult height is of 130cm (range, 120−145cm) in men and 125cm (range, 115−137cm) in women, based on specific standard growth curves.7,8 This stature is between 6 and 7 standard deviations (SDs) below the mean height in the population without achondroplasia.6
The skeletal manifestations give rise to other abnormalities: neurologic (foramen magnum stenosis, lumbar radiculopathy), otorhinolaryngolocial and/or psychological, among others.
Some studies show that FGFR3 is expressed in tissues other than bone, which suggests that the activity of FGFR3 could contribute to the extraskeletal manifestations of individuals with achondroplasia.
Therefore, achondroplasia requires multidisciplinary follow-up with the aim of preventing and managing complications, improve quality of life and promote autonomy and social inclusion of affected individuals.9,10
Different guidelines and recommendations have been developed to optimise the follow-up of achondroplasia, of which the most important are the recommendations of the American Academy of Pediatrics, published in 2005 and recently updated,9 the Japanese clinical practice guidelines,11 the first European consensus on principles of management for achondroplasia10 and the International Consensus Statement on the diagnosis, multidisciplinary management and lifelong care of individuals with achondroplasia.1 However, despite these recommendations, there is substantial variability worldwide in the management of individuals with achondroplasia, which has deleterious medical, functional and psychosocial consequences on the patients and their families.1
Therefore, these recommendations must be implemented in everyday clinical practice, adapting them to the specific circumstances of each health care system.
DiagnosisPrenatal diagnosisAbsent a positive family history, achondroplasia may be suspected from 22 weeks of gestation, although it is difficult to detect before 26 weeks. This usually results in delayed prenatal diagnosis in the third trimester, past the routine ultrasound scan performed at 20 weeks, when an ultrasound scan is performed for other reasons.
The sonographic features suggestive of achondroplasia are short limbed-short stature with rhizomelia, femoral bowing and deceleration of femur growth from 26 weeks, a femur length to foot length ratio of less than 1, brachydactyly with trident configuration of the hand, narrow chest cavity in the absence of pulmonary hypoplasia, distinctive facies (frontal bossing, midface hypoplasia and flat nasal bridge), square iliac wings and polyhydramnios.
Prenatal genetic testing is possible by chorionic villus sampling from 12 weeks or amniocentesis from 16 weeks if achondroplasia is strongly suspected in the first trimester, which would be the case of offspring of parents with achondroplasia or healthy parents with a previous child with achondroplasia. Non-invasive methods are currently being tried to detect pathogenic FGFR3 variants in foetal DNA extracted from maternal blood (in mothers without achondroplasia) as an alternative to amniocentesis, which, together with the ultrasound examination, can be safe and cost-effective non-invasive method for diagnosis of achondroplasia.12
A prenatal visit with a clinical geneticist and a paediatrician with expertise in achondroplasia is recommended and necessary once the condition is suspected in the foetus to provide accurate information. The professionals tasked with the initial delivery of the diagnosis and genetic counselling must be qualified to do so. These discussions must be held with both parents present, in an adequate setting and allotting an adequate amount of time. The providers must have up-to-date knowledge about available resources and the necessary training to develop a management approach. Involving representatives from patient associations in this process may be beneficial (http://gat-atenciontemprana.org/wp-content/uploads/2019/05/primera-noticia-web.pdf) (http://assets.comitedebioetica.es/files/documentacion/consejo-genetico-prenatal.pdf).
A delivery plan should be made, including the right setting and timing of delivery to manage any potential short-term complications in the infant. Coordinated care should be delivered by a multidisciplinary team including an obstetrician with expertise in achondroplasia, a geneticist, a neonatologist and a paediatrician. As regards delivery, an elective caesarean section is generally recommended due to the small pelvis and cephalopelvic disproportion, foetal macrocephaly and the risk of spinal compression during passage through the birth canal because of a relatively small foramen magnum.9
Clinical and radiological diagnosisClinical suspicion of achondroplasia in the newborn is based on the following features: microcephaly, broad, flat forehead, flat nose bridge, fingers in trident configuration, disproportionate short stature with rhizomelic limb shortening (proximal segments of humerus and femur disproportionately shorter than distal segments), redundant skin folds in extremities, long and narrow thorax and neonatal hypotonia.13
A full radiologic skeletal survey is not routinely required in the case of clinical suspicion of achondroplasia, but radiographs of the anteroposterior and lateral skull, lateral thoracolumbar spine, anteroposterior and lateral chest, abdomen and pelvis, anteroposterior of each upper and lower extremity long bone, and anteroposterior of the left hand should be obtained.1 The characteristic radiographic features of achondroplasia are a square shape of the pelvis with horizontal acetabula and a small sacrosciatic notch, short pedicles of the vertebrae with interpedicular narrowing from the lower thoracic through lumbar region, rhizomelic (proximal) shortening of the long bones, proximal femoral radiolucency, and a characteristic chevron shape of the distal femoral epiphyses, thoracolumbar kyphosis and shortening of the proximal and middle phalanges.9
Genetic diagnosisThe diagnosis of achondroplasia is based on clinical and imaging features, but genetic testing is necessary for molecular confirmation and reproductive genetic counselling.1
Ninety-nine percent of patients have the heterozygous c.1138G>A variant and 1% the c.1138G>C variant in the FGFR3 gene. The pattern of inheritance is autosomal dominant, with a penetrance of 100%. In 80% of cases, the disease is caused by a de novo variant, not inherited from the parents, and associated with advanced paternal age (generally>35 years). Achondroplasia is one of a small number of so-called RAMP disorders – recurrent, autosomal dominant, male biased, paternal age effect disorders – all of which likely arise because of their positive selective effect on spermatogonia. However, the risk of recurrence in the offspring of a healthy couple with an affected child is estimated at 1% due to the possibility of germline mosaicism.9 If one parent has achondroplasia, the probability of passing achondroplasia to the offspring is 50%, if both parents have it, there is a 25% probability of having a healthy child, a 50% probability of passing achondroplasia to the child and a 25% probability of the child having a lethal double-dominant form of the disease.
Pathogenic variants in the same gene cause diseases included in the differential diagnosis of achondroplasia, such as SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans) and hypochondroplasia (a disease similar a to achondroplasia, but with a milder phenotype).3
Genetic testing when clinical signs are present should be performed with targeted tests for the detection of the 2 variants of the FGFR3 gene associated with achondroplasia. In atypical presentations, next-generation sequencing techniques may be used, such as skeletal dysplasia gene panels, clinical exome sequencing or whole exome sequencing.1
Follow-upMultidisciplinary follow-up is required from diagnosis, as achondroplasia is associated with increased morbidity and potentially fatal complications and therefore affected patients may benefit from a scheduled follow-up plan. This multidisciplinary care should be delivered in a reference centre for skeletal dysplasias or a facility with providers with expertise in achondroplasia. The First European Consensus and the International Consensus recommendations, both published in 2021, underscore this aspect, especially for the first 2 years of life.1,10 The main goals of care are to anticipate, detect and manage complications and to support autonomy, quality of life and independence. Ongoing monitoring and expert management is also recommended through and beyond adolescence, including the transition to adulthood, genetic counselling and pregnancy management.10
The American Association of Pediatrics has published a timeline for the health supervision of people with achondroplasia through the lifespan (Table 1), with screening at each time point requiring anthropometric measurements, a physical examination, a neurological examination and an otorhinolaryngology evaluation. It is also important to watch for signs of complications in order to anticipate them.9 Patients with achondroplasia should undergo routine vaccination conforming to national immunization programmes.1
Timeline for the follow-up of patients with achondroplasia, adapted from the American Association of Pediatrics (2020).
| Birth-2 years | 2−13 years | Adolescence | Adulthood | |
|---|---|---|---|---|
| Growth (height/length, weight, head circumference) | X | X | X | X |
| Physical examination | X | X | X | X |
| Neurologic examination | X | X | X | X |
| Development | X | X | X | |
| Neuroimaging | X (between 6 months and 1 year or in presence of symptoms) | X (as indicated) | X (as indicated) | X (as indicated) |
| Polysomnography | X (before 1 year, preferably before 1 month) | X (as indicated) | X (as indicated) | X (as indicated) |
| Hearing/Ear, nose, throat assessment | X | X | X | X |
| Radiography for kyphosis, genu varus, bowing | X | X (as indicated) | X (as indicated) | X (as indicated) |
| Warning signs of potential complications | X | X | X | X |
| Obesity, exercise, diet | X | X | X | |
| Information about support group(s), family support | X | X | X | X |
| Genetic counselling | X | X |
Exhaustive monitoring is essential in this stage, if possible, every 2 o 4 months.1
Anthropometry and body proportionsThe anthropometric evaluation should include basic parameters such as weight, height, sitting height, head circumference and arm span. These measurements must be taken by trained professional and interpreted based on the growth charts of the individual patient and the comparison with specific tables for people with achondroplasia in the European population, of which the most recently published and currently recommended for follow-up are those of Neumeyer et al.,8 although other specific tables for the population with achondroplasia have been published in the past.7,14–16
Neurology/NeurosurgeryPatients with achondroplasia are at increased risk of neurologic complications because the cranial base develops by endochondral ossification and therefore the craniocervical junction anomalies are common. Complications may include foramen magnum stenosis, upper cervical vertebral canal stenosis, abnormal odontoid shape or position and ligamentous laxity in the cervical spine.17
The incidence of sudden death in the first year of life is 7.5% greater in individuals with achondroplasia compared to the general population, which seems to be due to chronic compression of the brainstem and spinal cord by constriction at the foramen magnum.17 There is also an increased risk of central sleep apnoea, although obstructive sleep apnoea is more frequent.18,19 Therefore, a polysomnography should be performed in the first year of life to rule out apnoeas that parents may have missed and any time there are symptoms of sleep apnoea.1 Some authors have proposed assessing neurologic risk by means of polysomnography, recommending its performance as early as possible, preferably 1 month post birth.9
A thorough neurological examination is essential to anticipate potential complications, in addition to neuroimaging tests if neurologic symptoms are present, along with exhaustive monitoring of any symptoms through age 2 years, as these complications are less frequent at later ages.1
Children with achondroplasia exhibit delayed milestone acquisition due to the particularities of their anatomy, but without involvement in other areas of development, so a delay in milestone attainment compared to the standard for achondroplasia could be indicative of a complication. There are adapted milestone charts for achondroplasia, among which we ought to highlight walking, achieved at a mean of 20 months (Fig. 1).9
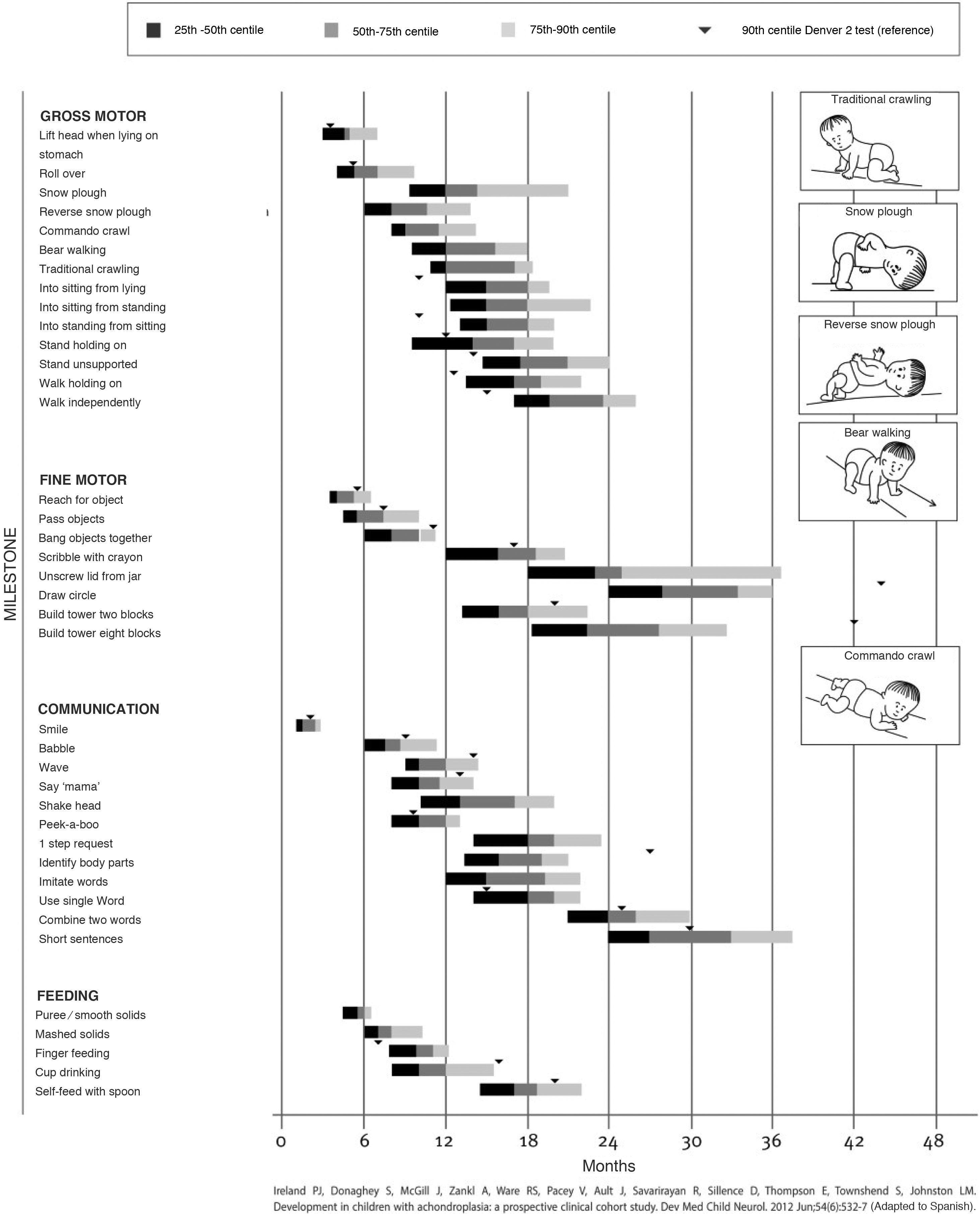
Tool for the assessment of development in children with achondroplasia developed by Ireland et al. (2012), reproduced with permission from the autor.40
Macrocephaly with excessive extra-axial fluid and asymptomatic ventriculomegaly are normal variants in achondroplasia that do not require referral. Hydrocephalus is a potential complication, but is rare.11
Monitoring of head circumference with measurements taken every 2–4 months is recommended in the first year of life. Excessive growth of the head circumference associated with any neurologic signs or symptoms is suggestive of craniocervical compression or hydrocephalus.1 It is important to examine the fontanelle and assess for the following warning signs and symptoms: difficulty swallowing, vomiting, hypertension, bradycardia, prolonged apnoea while sleeping, cyanosis, asymmetry of lower limb movements, decreased axial and peripheral tone, loss of acquired milestones, increased reflexes or early hand preference.1,9,11 It is important to avoid any head or neck injury, and use of adequate seats or carriers with cervical support is recommended.9 It is important to emphasise the need to avoid activities such as tumbling, trampoline jumping or high-impact, contact or collision sports in which the head could be hit.
As regards imaging tests, computed tomography (CT) can be used to compare the size of the foramen magnum with reference values and does not require sedation, but it does not allow investigation of the brainstem and upper spinal cord with the same definition as magnetic resonance imaging (MRI). Computed tomography is being replaced by MRI, with the drawback that the latter requires anaesthesia.20 Magnetic resonance imaging allows direct visualization of the brainstem and upper cervical spinal cord and can evince dynamic cord compression and alteration of cerebrospinal fluid (CSF) flow in achondroplasia, which is a better indicator of the need for surgical intervention.9 The first MRI-based classification for screening of foramen magnum stenosis was published in 2021, ranging from stage 0 (normal) to 4 (most severe).21
Although the review of the current guidelines did not allow us to make a clear recommendation, it is reasonable to deduce that performance of polysomnography as early as possible, even in the first month of life, is advisable for screening of neurologic risk. Imaging (CT or MRI based on availability, but preferably MRI) is recommended between ages 6 months and 1 year and in the presence of warning signs or symptoms.
The patient should be referred to a neurosurgeon in the presence of symptoms compatible with foramen magnum stenosis for consideration of surgical decompression, abnormal findings in the neurologic evaluation, unexplained poor weight gain, abnormal polysomnography results, imaging showing a small foramen magnum, substantial deformation of the upper cervical spinal cord, or lack of cerebrospinal fluid around the spinal cord.9 If surgery is required, it should be performed in a reference centre with a neurosurgery team with previous experience in the surgical management of children with achondroplasia1 and a qualified anaesthesia team (see the anaesthesia section below).
OtorhinolaryngologyPatients with achondroplasia are at increased risk of obstructive apnoea due to tonsil enlargement, glossoptosis and pharyngolaryngeal wall laxity and redundancy.18,19 Due to the anatomy of the ear canal, they are more likely to develop otitis during childhood and therefore to experience hearing loss, so it is important to conduct yearly ear, nose and throat evaluations. It is important to confirm that the newborn hearing screening was performed and to perform a full evaluation in infants with abnormal results.13 Whenever these children have an upper respiratory infection or exhibit irritability, a middle ear infection should be ruled out and, if present, should be promptly treated. Patients may exhibit language delays secondary to hearing loss,9 in which case referral to speech and language therapy is indicated.1
OrthopaedicsSpine: one of the most frequent problems in the early years of life is thoracolumbar kyphosis (90%–95%). Parents must be instructed to ensure adequate support to the back until muscle tone develops in the trunk, avoiding seats or carriers with insufficient support. This may require early assistance from an occupational therapist or rehabilitation specialist. The back and head should be aligned during feedings, supported by firm pillows. Mild thoracolumbar kyphosis usually improves once the child starts to walk; moderate kyphosis (20–40 degrees) a year after walking starts requires an orthopaedic evaluation and very rarely requires surgery. Children with persistent kyphosis (10%) are at increased risk of lumbar stenosis/neuropathy at later ages.1,9,13
Extremities: the main features are hypotonia and joint hypermobility, which make tripping and falling more likely when the child starts to walk, something parents must be informed of so they take appropriate precautions while still encouraging the child to be active.1 Another common feature is the external rotation of the hips, which usually disappears spontaneously when the child begins to bear weight.9 These children may exhibit peculiar crawling patterns that are not pathological.
AnaesthesiaIt is particularly important that the provider delivering anaesthesia have experience with achondroplasia, as these patients require careful control of cervical movement (difficult airway and risk of craniocervical compression). Furthermore, anaesthetics must be dosed based on body weight, not age, and it may be difficult to establish venous access because of incomplete elbow extension. Lastly, spinal or epidural anaesthesia should be avoided unless previous neuroimaging reveals adequate space inside the spinal canal and there are no signs of neurologic compromise.
Follow-up from age 2 to 13 yearsIn this stage, evaluations are recommended every 6–12 months, choosing the appropriate interval on a case-by-case basis.1
AnthropometryRecommended in each appointment.
Neurology/NeurosurgeryOngoing monitoring for signs and symptoms of cervicomedullary compression is recommended.
Delayed fontanelle closure is common, usually occurring at age 5–6 years.
OrthopaedicsSpine: ongoing monitoring of thoracolumbar kyphosis and lumber hyperlordosis is recommended, completing the follow-up with a neurological examination.
Upper extremity: the range of motion of the elbows must be monitored on account of the characteristic limited flexion.
Lower extremities: genu varum with tibial torsion is frequent, affecting 40%–70% of patients.22,23 This deformity can cause symptoms, including mechanical pain, impaired gait and limitations to physical activity. Thus, the follow-up protocol must include periodic comprehensive clinical evaluations of lower limb alignment in prone, supine and standing positions as well as during gait to analyse internal tibial torsion, mediolateral instability and genu varus and recurvatum.1 If the deformity is symptomatic and/or progressive, a radiological evaluation should also be performed, obtaining anterior–posterior and lateral weight-bearing standing radiographs of the full length of both legs. Bracing is not indicated for treatment of this deformity.1
Otorhinolaryngology/PulmonologyLongitudinal monitoring of hearing, ear infections and language development is recommended. If the patient requires myringotomy, the specialist should look for otoscopic signs of a high jugular bulb, as it increases the risk of damage during the procedure.24 Continued monitoring of apnoeas is also necessary during this period, and tonsillectomy should be considered in patients with obstructive apnoea due to enlarged tonsils, although this is not the only contributing factor (achondroplasia manifest with particular craniofacial features) and therefore surgery does not always achieve resolution of apnoea.19 In patients exhibiting language delays past age 2 years, hearing loss should be ruled out.
Patients with suspected obstructive apnoea also require assessment by a pulmonologist. Restrictive pulmonary disease is infrequent, and obstructive disease is more common. Gastro-oesophageal reflux disease may be present, especially in patients with neurorespiratory complications.9
ObesityObesity was a frequent comorbidity even before it became a global epidemic, so it has been hypothesised that achondroplasia may intrinsically facilitate its development. Obesity develops early with preferential abdominal fat accumulation. These patients have limitations to physical activity, a tendency to eat constantly and a lower basal metabolism compared to people of average height of the same age and sex (although higher than expected for their height and weight), and it seems that the FGFR3 variant itself promotes the differentiation of mesenchymal stem cells into adipocytes.25 Prevention and monitoring of weight and the body mass index in every visit using specific tables for achondroplasia26 are important, as obesity worsens neurologic and orthopaedic problems and apnoea and increases the likelihood of cardiovascular disease, the leading cause of death in individuals with achondroplasia, at earlier ages. Obesity is managed with personalised dietary and physical activity measures1 and psychological support if needed.25
Early interventionEarly intervention refers to the set of interventions aimed at children aged 0–6 years, their families and their environment to address any current or potential future temporary or permanent needs as early as possible. The coordination of specialists from different fields is necessary for delivery of early intervention. It requires an effective coordination of services, fluid communication and active involvement of every professional directly or indirectly working with the patient. It also requires the interdisciplinary coordination of the health care, education, social work and early intervention systems as well as patient associations.
Early intervention services for children with achondroplasia should address all areas of development from an early stage. Also important are the initial assessment and assessment of patient progress in the most important areas that may be affected, such as gross and fine motor skills, language and communication skills, cognition, autonomy and interpersonal relationships (https://www.infad.eu/wp-content/uploads/ConOtraMirada_guia_acondroplasia.pdf).
- 1
Gross motor skills: intervention in this area consist of physical therapy with 2 main goals, namely, achieving age-appropriate gross motor skills and preventing potential complications. Specific considerations should be taken into account, such as avoiding crawling. When the patient starts to walk, given the risk of lumbar hyperlordosis, it is important to avoid the prone position with an elevated head. The patient must be trained to move from the floor to standing correctly, avoiding knee hyperextension while changing positions.
- 2
Language and communication: severe language impairments are not common in children with achondroplasia, but language acquisition should be closely monitored, and delays past age 2 years should be considered a warning sign of potential undiagnosed hearing loss. Intervention in this area consists of speech and language therapy with the general goals of improving sound articulation and achieving adequate expression and communication. Specific goals should address potential respiratory complications due the narrowing of the nasal cavity and fine motor skills in the speech organs to achieve adequate pronunciation of specific phonemes and normal chewing and swallowing. Taking into account specific features such as prognathism, high-vaulted palate or macroglossia, which result in malocclusion, restricted tongue movement and therefore difficulty articulating and pronouncing certain sounds. With the goal of achieving adequate speech and hearing, patients work on skills related to breathing, blowing, swallowing, auditory discrimination, orofacial motricity, articulation, intonation, stress and rhythm.
- 3
Fine motor skills: the general goals in this area are to improve in-hand manipulation and eye-hand coordination. Specific goals are to achieve age-appropriate fine motor skills, promoting the development of the pincer grasp and graphomotricity. Also, necessary adaptations will be contemplated, such as the use of a shorter pencil, adaptors, smaller scissors, wide-format paper, etc.
- 4
Cognition: cognition is not usually impaired in achondroplasia, but it is important to take into account that delays in other areas of development may affect the development of cognitive skills. Possible determinants of cognitive development include gross motor delay in the first years of life, fine motor impairment, delayed speech, poor self-image and impaired autonomy. Interventions for cognitive development can be integrated in interventions addressing the other areas.
- 5
Autonomy and interpersonal skills: The general goals are the progressive acquisition of self-competence and a positive and healthy self-image. Specific goals include the development of confidence in the ability to adapt to the environment, acquiring coordination and motor control in play and activities of daily living, using any necessary adaptations to improve autonomy.
Dental assessments by an odontologist are necessary from an early age, and patients frequently require orthodontic treatment.24
Adolescence-early adulthoodIn this stage, follow-up evaluations are personalised.1
The follow-up of patients requires assessments of metabolism (obesity) and oral health, monitoring of ear, nose and throat problems (apnoeas, hearing) and orthopaedic evaluations. From a neurological standpoint, they may develop signs or symptoms of nerve compression due to spinal stenosis, so it is important to check reflexes, tone and sensory findings and assess for incontinence. It is also important to assess chronic pain, which is frequent in patients with achondroplasia.1,9
Genetic counselling should be provided, informing all patients about contraception and discussing prenatal testing, pregnancy and its potential complications (restrictive lung disease, cardiovascular disease, complications of anaesthesia) and the recommendation of caesarean delivery with female patients·9
AdulthoodLife expectancy may be reduced, with a 10-year earlier mortality on account of the increased risk of cardiovascular disease.1,25 Functioning and health may deteriorate considerably in the fourth decade of life. Quality of life can decrease due to functional impairment and psychosocial challenges, and nearly 50%–60% have low self-esteem and anxiety problems.10
Psychological supportFrom the time of diagnosis, either prenatal or postnatal, psychological support must be offered, as well as information on the management of the child, including expectations for the future, and on associations and support groups, which are a significant source of support for the family.1,10 This process should also include siblings of patients to help address any questions, concerns or conflicts that arise in the family.1 During childhood and adolescence, functional evaluations should be performed periodically to identify limitations in walking, self-care and daily functioning, providing any necessary means to achieve independence. As the child grows, adaptations need to be made to the home, school, bathroom and car seat to promote autonomy. Some adolescents may avoid using adapted tools in school out of concern of being mocked by their peers or being asked uncomfortable questions, so they must receive support to address these obstacles, and peer support groups may offer essential assistance at this age.1 Physical and occupational therapists play key roles in achondroplasia.1,9 Quality of life is reduced in both patients and their parents compared to healthy peers of the same age and their families, and efforts must be made to improve it.27 As is the case with other adolescents, drug, alcohol and tobacco use and sexual activity should be discussed, in addition to reproduction and contraception, expectations for the future and the need for adapted vehicles, and psychological support is of the essence.9
TreatmentSurgeryMost surgical interventions are performed to manage complications. The most frequent type of procedures in the early years of life are neurosurgery (foramen magnum stenosis and ventriculoperitoneal shunts), ear, nose and throat surgery (myringotomy, tonsillectomy and adenoidectomy) and, less frequently, orthopaedic surgery for treatment of thoracolumbar kyphosis. In adulthood, surgical procedures are most frequently performed to manage lumbar spinal stenosis.
We also ought to mention orthopaedic surgeries for limb lengthening and correction of genu varus in the management of the skeletal features of achondroplasia.
As regards genu varus, the indication of surgery is established based on the severity of the deformity, the associated symptoms and the resulting functional impairment, and different approaches have been described, including osteotomy and the use of guided growth plates.
Different approaches have been described for lengthening the lower limbs (femur and tibia) and upper limbs (humerus), with its indication and the appropriate age at initiation, surgical technique and expected outcomes subject to ongoing debate, and a high incidence of complications.9,11 Lower limb lengthening (by 14−40cm) aims at increasing the length of the lower extremities, thereby improving the standing weight, body proportions, functioning and quality of life that depend on the lower segment of the body. Similarly, upper limb lengthening (by 8−10cm) would increase the length of the arms and arm span, improving the body proportions, function and quality of life that depend on the upper segment of the body.28–31
The different surgical approaches are usually described independently as opposed to integrated in a care bundle with clearly defined objectives. The surgical management of these patients requires an integral approach, be performed in centres with experience in the management of achondroplasia and with a multidisciplinary approach to decision-making before and after the procedure that will take into account anthropometric, radiological, functional and quality of life-related variables.
Since effective medical treatment has only recently become available and while data on the long-term outcomes of drugs that are currently authorised, undergoing trials or in development, surgical management is a valid approach for limb lengthening and correction of deformities in a specific subset of patients with achondroplasia that remains to be clearly defined. Long-term outcomes of medical treatment also need to be investigated to determine whether surgery will continue to be the sole therapeutic option, be considered an adjuvant to medical treatment or should no longer be used.
Medical treatmentGrowth hormone therapy has been approved in Japan,11 but there is no evidence that it has a relevant effect and long-term body proportion outcomes are not known.1,13 This approach is no longer indicated.
At present, drugs that target the FGFR3 receptor are being developed to block its activation, inhibit its intracellular signalling pathway or increase its turnover.5 Among them, we ought to highlight the effects of a recently discovered C-type natriuretic peptide analogue (vosoritide) with a longer half-life than the endogenous growth factor. On binding its receptor, it inhibits the MAPK pathway (activated by the product of the pathogenic variant of the FGFR3 gene) at the level of RAF-1, increasing chondrocyte proliferation and differentiation at the epiphysis. In 2021, the European Medicines Agency (EMA) authorised vosoritide (Voxzogo) for treatment of patients with achondroplasia confirmed by genetic testing aged 2 or more years and before epiphyseal closure, to be administered once a day by subcutaneous injection. The height and weight of the patient must be monitored during treatment with vosoritide. Treatment should be discontinued if linear growth velocity exceeds 1.5cm per year and/or once epiphyseal closure occurs. There is no data on the safety and efficacy of vosoritide in patients with kidney or liver failure.
An initial phase II trial in 35 patients aged 5–14 years (BMN 111-202; EudraCT number 2013-004137-32) established that the optimal dose in terms of efficacy and safety was 15μg/kg/day, without significant increases in the effect of the drug at higher doses.32 The 15μg/kg/day dose was later used in a phase III double-blind randomised controlled trial (BMN 111-301; EudraCT number, 2015-003836-11) in a total of 121 patients aged 5–18 years randomly allocated (1:1) to vosoritide or placebo for 52 weeks of treatment, which found an increase in the annualized growth velocity of 1.57cm/year (95% confidence interval [CI], 1.22–1.93) with the use of vosoritide compared to placebo (P<.0001).33 The phase III extension study continued treatment for a cumulative duration of 104 weeks (BMN 111-302; EudraCT number 2017-002404-28; NCT03424018), with administration of vosoritide to all patients at a dose of 15μg/kg/day, and found that the increase in linear growth continued during the second year of treatment with no evidence of tachyphylaxis, with a mean annualized growth velocity in week 104 of 5.52cm/year (standard deviation [SD], 1.77) in patients treated with vosoritide from the outset and of 5.43cm/year (SD, 2.03) in patients switched from placebo to vosoritide. Comparing the group treated with vosoritide with the untreated group, the observed difference in height in year 1 of treatment, 1.73cm, was similar to the difference observed in year 2, 1.79cm. The additional height gain achieved in the 2-year treatment period was of 3.52cm more compared to untreated children. It seems that once vosoritide achieves its maximum effect, it remains constant through time, with a height z-score of +0.44 (95% CI, 0.25−0.63) in week 104. There was also an improvement in body proportions, with a decrease of –0.05 in the upper-to-lower body segment ratio (95% CI, –0.09 to –0.01) compared to untreated individuals. The most frequent adverse events were mild and transient injection site reactions, vomiting and transient decreases in blood pressure that were usually asymptomatic and resolved spontaneously. There were a total of 14 adverse events in the trial, but none was attributed to the drug, and there were no adverse events related to disproportionate bone growth or bone disease.34
Currently 2 other clinical trials of vosoritide are underway. The first one is a phase II clinical trial (BMN 111-206, EudraCT number 2016-003826-18, NCT03583697) to assess the safety and efficacy of vosoritide in children aged 0–60 months. The second is a phase II trial in 20 patients aged 0–12 years (BMN 111-209, EudraCT number 2020-001055-40, NCT04554940) to assess whether early administration in patients at risk of cervicomedullary compression that could require surgery is efficacious and safe.35
Other drugs in the development pipeline include another natriuretic peptide (TransCon CNP) with a longer half-life, currently in phase II trials (NCT04085523), that is administered subcutaneously once a week in children weekly via the subcutaneous route in children with achondroplasia aged 2–10 years.36,37
Another molecule, soluble FGFR3 (recifercept), used as a ligand of FGF to prevent its binding of FGFR3 is currently under study in a phase II trial (NCT04638153) that is showing promising results both in terms of skeletal deformities and obesity with early administration.37,38
Yet another molecule under investigation, initially developed for cancer treatment, is an orally administered tyrosine kinase inhibitor of FGFR3 (infigratinib) that blocks the intracellular signalling pathways of FGFR family receptors. In a mouse model, it improved the growth of all four limbs and mandibular abnormalities and increased the size of the foramen magnum. A phase II trial is currently underway (NCT04265651) in previous participants in the PROpel 2 study, and is expected to be completed in 2026.37,39
We await evidence to be able to assess the long-term efficacy and safety of these drugs and their potential synergistic interactions. A recent publication explored the advantages and disadvantages of treatments under investigation to analyse the impact each of them has on the comorbidities associated with achondroplasia.37
ConclusionAchondroplasia is a rare disease and is the most frequent type of skeletal dysplasia manifesting with short stature. In addition to disproportional short stature, it has neurologic, otorhinolaryngologic, orthopaedic, endocrine, and psychosocial manifestations, and therefore requires multidisciplinary management to prevent and treat its complications. It is essential for affected individuals to receive psychological and peer support to improve quality of life and promote autonomy. New treatments are currently being developed, and vosoritide, based on the efficacy and safety results found in clinical trials, has been authorised by the EMA for treatment of patients with achondroplasia aged 2 or more years who have not yet stopped growing. Real-world studies will allow us to assess its impact on body proportions, function and quality of life, among other outcomes. It is also important to keep abreast of efficacy and safety results for other drugs in development to gain a comprehensive perspective of future therapeutic options in achondroplasia.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors wish to thank Fundación ALPE, especially Carmen Alonso and Susana Noval for their invaluable collaboration.
