


La incontinencia pigmenti (IP) es un trastorno neurocutáneo raro, con una frecuencia de 1 en 50.000 recién nacidos, de etiología genética asociada a mutaciones en el gen IKBKG (NEMO) en Xq28, con herencia dominante ligada al X. Tiene una presentación clínica de manifestaciones muy variables detectadas desde la etapa neonatal, con 3 estadios bien definidos en forma secuencial, solapada o salteada, y cada una de estos con un diagnóstico diferencial distinto. Mediante la técnica molecular de PCR+RFLP se analizó el gen IKBKG en cuatro pacientes diferentes con manifestaciones sospechosas de IP además de la biopsia de piel confirmatoria; en todas se detectó la deleción de los exones 4 al 10. Destacamos que ante la sospecha clínica de IP es importante el estudio familiar y el multidisciplinario (complicaciones neurológicas, oculares…), y el necesario asesoramiento genético.
Incontinentia pigmenti (IP) is a rare neurocutaneous disorder with a frequency of 1 in 50,000 newborn, and is associated with mutations in IKBKG gene (NEMO) in Xq28, inherited as an X-linked dominant trait. Clinical manifestations detected since the newborn period are highly variable, with 3 well established sequential or overlapped states and each with a characteristic differential diagnosis. With PCR+RFLPs, we analyzed the IKBKG gene in 4 patients with different clinical manifestations and characteristic skin biopsy. In all 4 patients the same deletion of exons 4 to 10 was identified. In female patients in whom the dermatological lesions lead to the suspicion of an IP diagnosis, it is important to have the complete, multidisciplinary and molecular analysis of their first level female relatives. This should give us a clear diagnosis, which is the first step to complete genetic counselling.
La incontinencia pigmenti (IP) es un trastorno neurocutáneo raro que se expresa casi exclusivamente en el sexo femenino. Fue descrito por primera vez por Garrod en 1906, definido posteriormente por Bloch en 19261 y por Sulzberg en 1928, respecto a sus características clínicas e histopatológicas2.
De etiología genética dominante ligada al X, con expresión variable en los tejidos derivados del neuroectodermo y mesodermo, su frecuencia es de 1 en 50.000 recién nacidos3, aunque probablemente sea mayor porque puede pasar fácilmente desapercibida al ser su presentación muy variable y las lesiones, posiblemente confundidas con las de otras entidades: infecciones virales o bacterianas, reacciones como el eritema tóxico, máxime en el periodo neonatal y en las etapas iniciales4.
La afectación cutánea evoluciona en tres estadios: 1° eritemato-bulboso o vesiculoso, 2° verruco-liquenoide y 3° pigmentario5. Puede observarse en algunos casos un 4° estadio con lesiones hipopigmentadas, atróficas, alopecia cicatrizal y alteración de uñas, considerado la manifestación en edad adulta de la IP6.
Generalmente estos estadios se presentan en forma cronológica aunque los signos iniciales pueden estar presentes en el momento del nacimiento o bien aparecer en las primeras 2semanas de vida, aunque es posible que los primeros estadios inflamatorios ocurran intraútero, así como que no progresen en la vida extrauterina de manera secuencial, o incluso, que no se presenten algunas de las fases en todas las pacientes7. Es más, los tres primeros estadios pueden coexistir y también estar ausente uno de ellos8.
Presentamos aquí cuatro pacientes con diferentes manifestaciones clínicas y evolución, lo que refleja su expresividad tan variable.
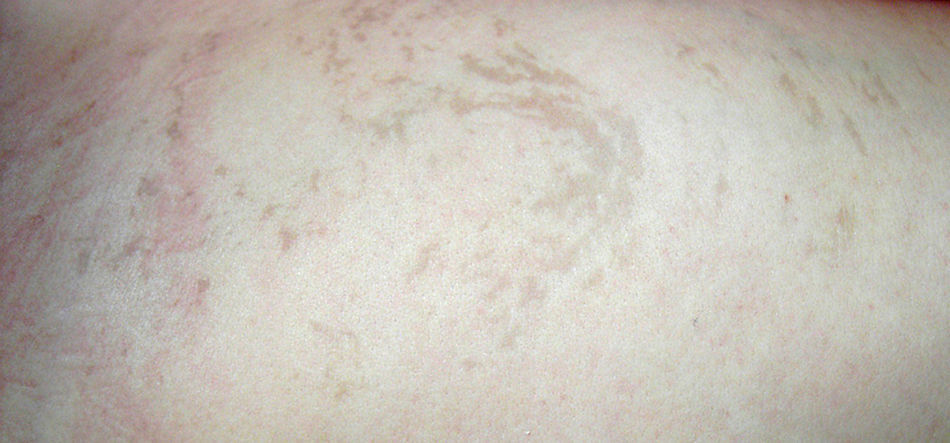
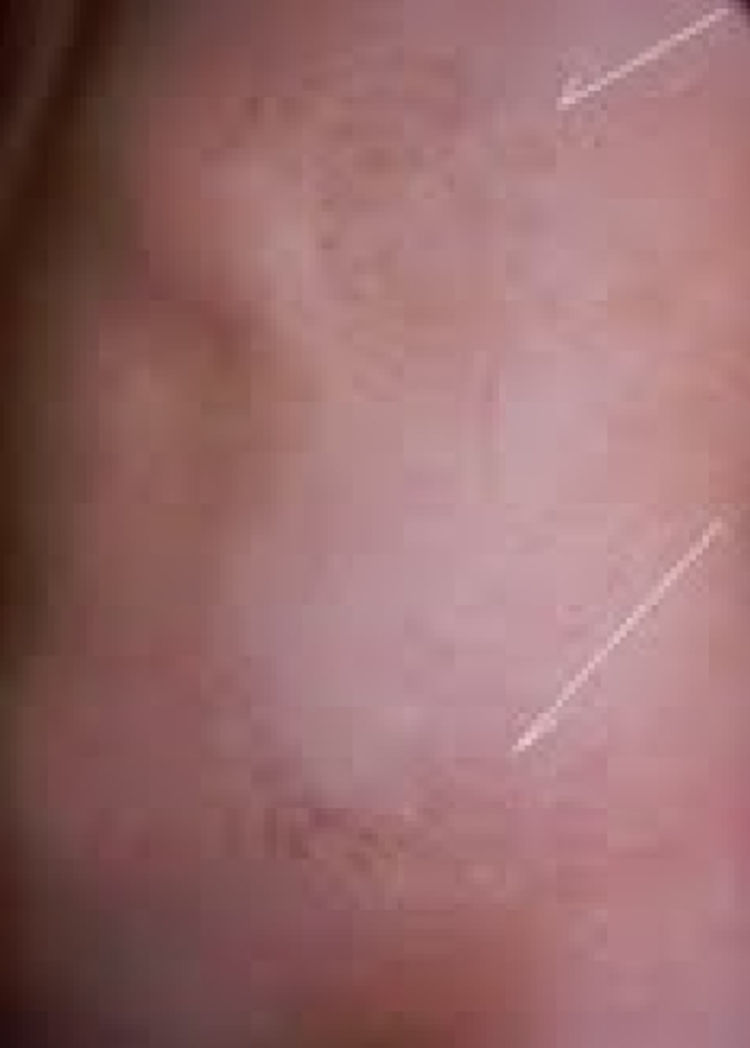
Casos clínicosCaso 1Niña de 8meses en la primera consulta, hija única de padres jóvenes, no consanguíneos, sin antecedentes familiares de importancia. Nació por cesárea por sufrimiento fetal agudo, a las 40semanas de gestación, con un Apgar 9-10, eutrófica y presencia de manchas hiperpigmentadas de predominio en tronco, las cuales se mantienen hasta la edad de la primera consulta en Neuropediatría, por asociar hemiparesia, hipertonía, disminución de la fuerza y movilidad de extremidades derechas, con clonus plantar y rotuliano; manchas espiroideas, hipermelánicas, en cara anterior de abdomen, región perigenital, tórax (región peri-areolar) y axilas (fig. 1). Crecimiento acorde con edad y sexo, hasta los 17meses (p90-97 en longitud, peso y perímetro cefálico); en esta edad, dirige la mirada, hay sonrisa social y atiende a su nombre, pero no hay sedestación.
Estudios complementarios
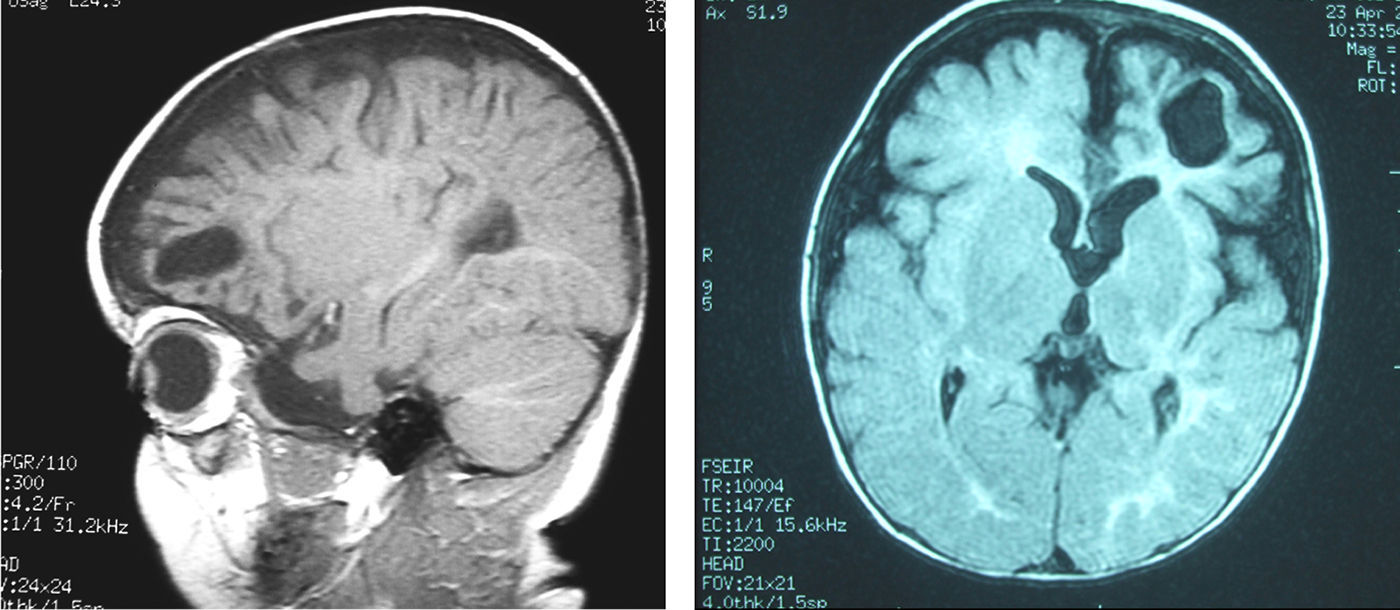
EEG con lentificación de la actividad de base en la región temporal izquierda. Resonancia magnética cerebral: atrofia córtico- subcortical e hipoplasia del cuerpo calloso, porencefalia frontal izquierda y lesión isquémica parietooccipital derecha con afectación difusa de la sustancia blanca (fig. 2).

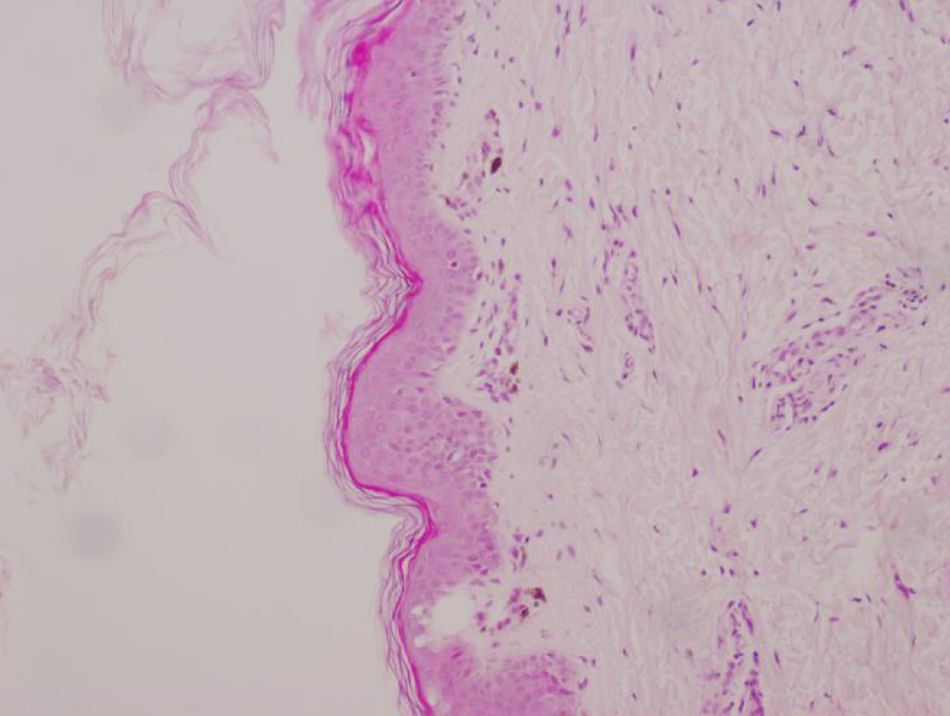
Cariotipo en sangre periférica y en fibroblastos de piel melánica: 46,XX. Biopsia de piel hiperpigmentada, con hallazgos que apoyan la sospecha clínica de IP (fig. 3).

Estudios de reordenamientos subteloméricos por MLPA normales, PCR y RFLP de gen IKBKG (Balagué Center) (BC) con deleción de los exones 4 a 10, identificándose como mutación de novo (los padres con estudio molecular normal).
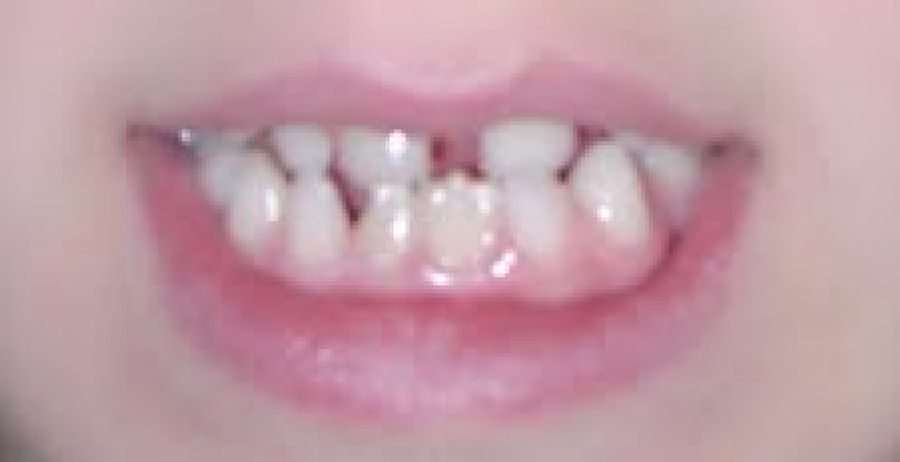
Caso 2Niña de 11años, tercera hija de madre de 44 y padre de 46años, sanos, no consanguíneos. Por el antecedente de edad materna avanzada se realiza amniocentesis citogenética sin complicaciones y resultado 46,XX. Al nacimiento se aprecian lesiones vesículo-ampollosas de predominio en región proximal de extremidades superiores y en inferiores, que evolucionaron a induradas y amarillentas. Crisis convulsivas mioclónicas de predominio en hemicuerpo izquierdo. Ecografía transfontanelar neonatal, normal. A los 3años, con desarrollo psicomotor normal, presentaba lesiones hiperpigmentadas y algunas zonas depigmentadas, cejas escasas y anodoncia parcial (fig. 4). Las lesiones de piel han evolucionado a hipercrómicas en hemiabdomen izquierdo y en tórax (fig. 5).
Estudios complementarios

En la biopsia de piel se describe ampolla multilocular intradérmica asociada a espongiosis con numerosos eosinófilos intravesiculares y dérmicos.
Estudio molecular con PCR+RFLP de gen IKBKG (BC): deleción de los exones 4 a 10, que no se detecta en el ADN de la madre.
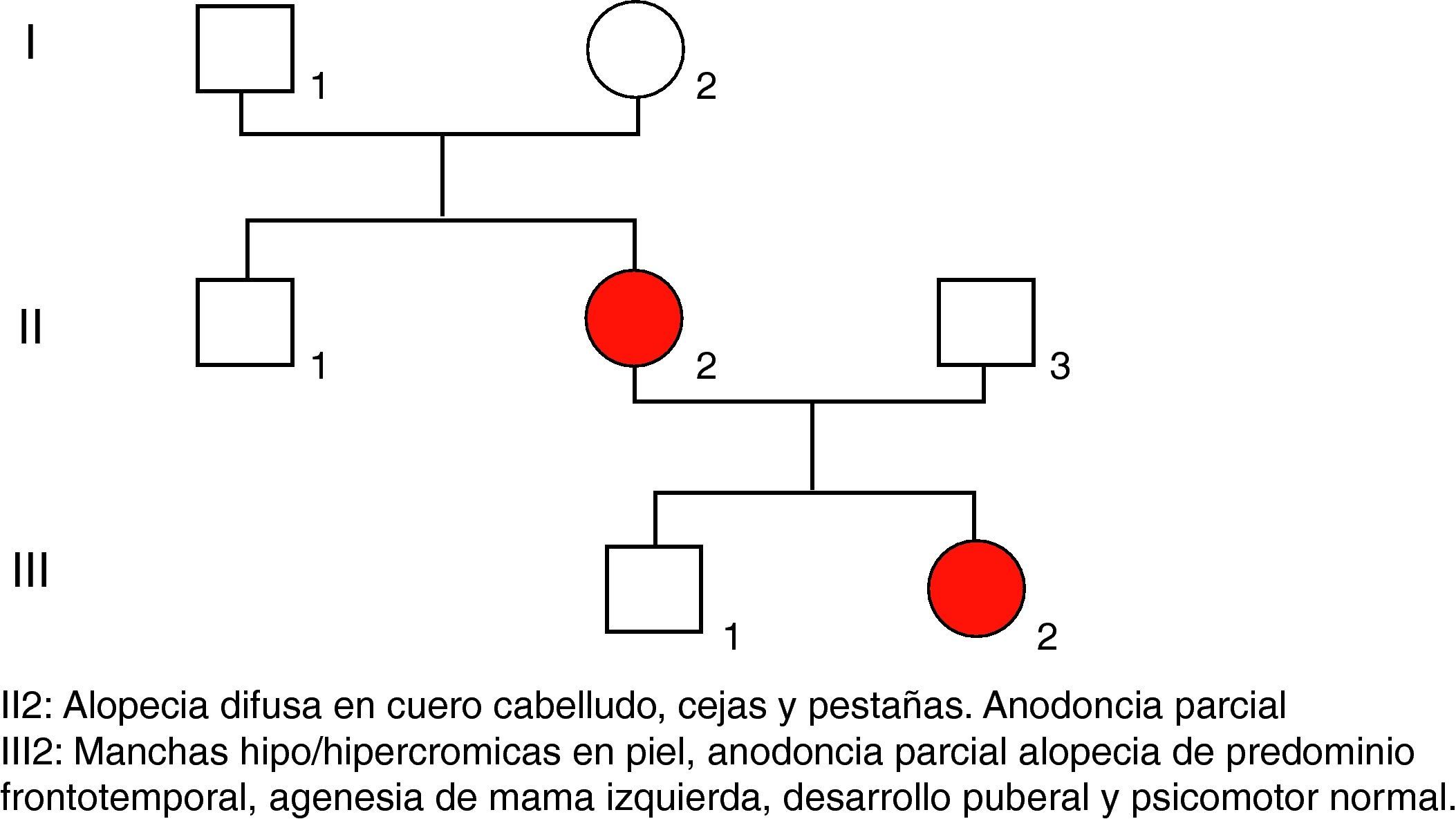
Caso 3Adolescente de 16 años, conocida desde los 2años, hija de padres jóvenes, no consanguíneos, sanos, al igual que el hermano mayor (fig. 6, árbol genealógico).

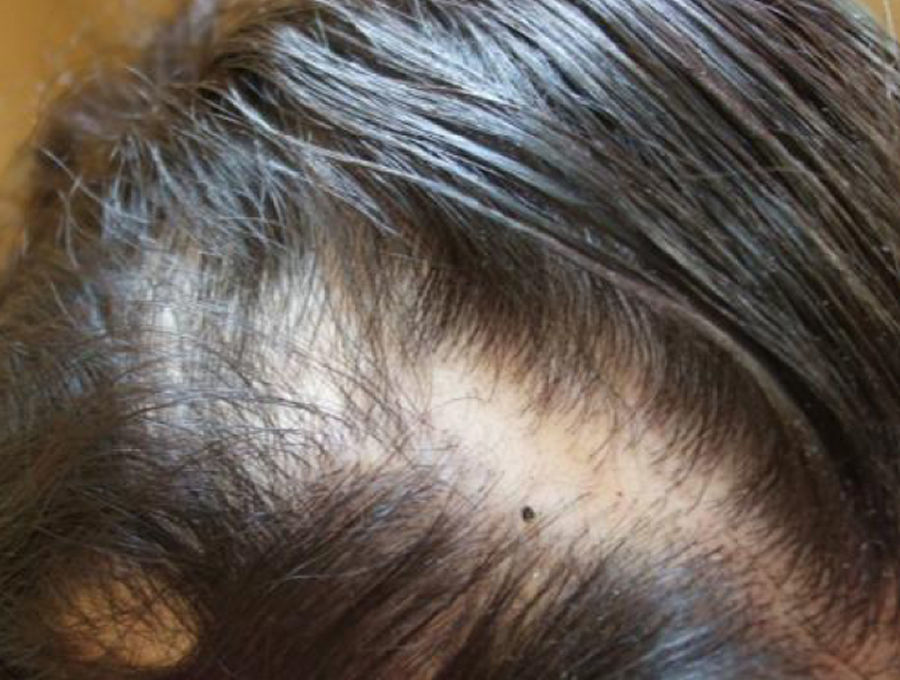
Tiene el antecedente neonatal, en otro hospital, de presencia de múltiples vesículas y lesiones ampollosas, e imágenes de aspecto verrucoso y disposición lineal. La biopsia de piel caracteriza el diagnóstico de IP. A los 2años y 3meses las lesiones se describen como manchas hipocrómicas diseminadas, algunas lineales, sin alteraciones tróficas. Posteriormente, zonas de alopecia difusa en cuero cabelludo, agenesia de piezas dentarias con diastema importante entre incisivos centrales superiores, y formas anómalas como dientes conoides y cúspides accesorias (fig. 7). Mamila izquierda significativamente menor que la contralateral y pezón accesorio del mismo lado. Parámetros de crecimiento y desarrollo psicomotor dentro de lo normal para su edad y sexo. Menarquia a los 13años y desarrollo puberal normal. Se ofrece corrección quirúrgica de asimetría mamaria.
Caso 4
Madre de caso 3, la cual presenta zonas de hipo e hiperpigmentación en piel, alopecia difusa (fig. 7), cejas y pestañas escasas y anodoncia parcial.
En ambas se hace estudio molecular mediante PCR+RFLP (BC) del gen IKBKG que mostró la deleción de los exones 4 a 10.
Se ofrece asesoramiento genético familiar como entidad dominante ligada al X con expresividad variable, y un riesgo de 50% de hijas afectadas y 50% de hijos varones con lesiones mucho más graves, generalmente letales en etapas tempranas, en la descendencia de ambas.
DiscusiónEn pacientes femeninas con manifestaciones cutáneas y sistémicas que puedan encajar en el diagnóstico de IP, hay que valorar los antecedentes familiares y personales, la evolución de las lesiones y las características de éstas, y la biopsia de piel hiperpigmentada (con datos histológicos característicos, bien descritos por Rola et al, también por Nso-Roca et al8,9).
Los criterios clínicos propuestos por Landy y Donnai en 199310 aún siguen utilizándose como guía para el diagnóstico clínico de la IP (tabla 1).
Criterios clínicos de incontinencia pigmenti según Landy y Donnai (1993)
| Grupo 1. Sin familiares afectados | Grupo 2. Con familiares afectados |
| Criterios mayores (al menos 1)Rash neonatal: eritema/vesículaeosinofiliaHiperpigmentación de lesiones anteriores(líneas de Blaschco)Lesiones alopécicaslinealesatróficasCriterios menores (apoyo a mayores)Alteraciones dentales: hipo-anodonciadientes cónicosretraso erupciónAlteraciones uñas/peloAlteraciones oftalmológicas:retinacataratasmicroftalmíanistagmus | Un familiar de primer grado afectadoUna o más de las siguientes:Piel.- hiperpigmentacióncicatrices hipopigmentadaslesiones alopécicasAntecedente de rash neonatal típicoDentición anómalaEnfermedad retinianaPérdidas gestacionales recurrentesde fetos masculinos. |
La IP se asocia a manifestaciones extradermatológicas en el 50 al 80% de los casos, y afectan predominantemente a tejidos derivados del neuroectodermo y de la cresta neural. Incluyen: afectación dentaria en el 90% de pacientes, con la alteración del esmalte como la más frecuente11. Defectos esqueléticos como deformidades craneales, espina bífida o fisura palatina en el 40%12, lo mismo que las retinianas: desprendimientos, proliferaciones fibrovasculares o cambios pigmentarios6,13,14.
Las alteraciones neurológicas son frecuentes: en el 13 al 50% de las pacientes y cobran importancia dado que su hallazgo marca el pronóstico de la enfermedad en cuanto al desarrollo psicomotor y futuro neurológico. Pueden expresarse con datos clínicos de epilepsia, retraso psicomotor, parálisis espástica o mixta, como en el caso 1, o sin repercusión clínica aparente3,9,13,15. Otras como la hipoplasia mamaria (caso 3) es citada como 10 veces más frecuente que en la población general10.
La presencia de todos estos datos extracutáneos es importante ya que pueden plantear el diagnóstico diferencial, dependiendo del estadio de la IP, como sigue: los 1.° y 2.° pueden confundirse con varicela zoster o candidiasis, sífilis congénita, impétigo ampolloso estafilocócico y dermatitis herpetiforme del neonato, que no mejoran con los tratamientos específicos y habituales9. En estadios más avanzados hay que descartar otros diagnósticos frecuentes en la población, aunque también raros como la condrodisplasia punctata ligada al X, hipoplasia dérmica focal, hipomelanosis de Ito, epidermólisis bullosa y la hipermelanosis nevoide espiroidea y lineal7 como en el caso 1.
Las manifestaciones de la enfermedad son las del gen alterado que se expresa en las células de un tejido de manera predominante aunque no única. Debido a que este gen se localiza en el cromosoma X, el cual tiene una expresión diferencial en cada célula, tenemos aquí también un mosaicismo de función, y por ello con una gran variabilidad clínica aun dentro de una misma familia con varios miembros afectados. Se trata por tanto de un mosaicismo génico que a su vez origina uno funcional.
El producto del gen IKBKG tiene como función normal la regulación de la expresión de otros genes que controlan al sistema inmune, la inflamación y la protección de apoptosis. En las pacientes afectadas existe una ausencia de activación de NFkB que se expresa como una alta sensibilidad a la apoptosis inducida por el factor de necrosis tumoral alfa15. La progresión de la enfermedad es causada por la muerte selectiva de las células que presentan la mutación en IKBKG (NFkB essential modulator), localizado en el brazo largo del cromosoma X (Xq28), con una deleción de los exones 4 a 10 en el 80% de las pacientes con IP5,7, pero también se han descrito otras que causan proteínas truncadas, no funcionales o sustituciones de nucleótidos16,17. Se han encontrado mutaciones en IKBKG en otras enfermedades como la displasia ectodérmica anhidrótica (DEA) con o sin inmunodeficiencia, con o sin linfedema, y también en osteopetrosis5. Parece ser un gen esencial para la viabilidad ya que los varones hemicigotos afectados mueren intraútero (en la mayoría de los casos) y las mujeres sobrevivientes presentan el cromosoma X mutado, preferencialmente inactivado18, en los linfocitos periféricos del 92% de ellas19.
La IP se transmite de forma dominante ligada al X, con una penetrancia del 100% pero con expresión altamente variable, lo cual queda de manifiesto en las cuatro pacientes que aquí presentamos, ya que todas presentan la misma deleción, siendo las manifestaciones clínicas, su inicio de presentación y evolución muy distintas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

