
















Los hipercrecimientos somáticos conforman una patología compleja, heterogénea y conocida parcialmente, si bien el incremento en nuestros conocimientos en biología molecular está posibilitando descubrir las bases etiológicas de muchos de los cuadros clínicos responsables. El diagnóstico diferencial de un paciente con una posible variante de la normalidad, una cromosomopatía, un síndrome dismórfico, una metabolopatía o una endocrinopatía, es esencial. La aproximación clínica inicial debe incluir una correcta anamnesis y examen físico, así como la solicitud de unas pruebas complementarias analíticas y de imagen que ayuden a orientar el diagnóstico. En efecto, es necesario practicar hemograma y bioquímica completos, determinar los niveles de IGF-I e IGFBP-3, T4 libre, TSH y homocistinuria, así como efectuar un cariotipo y una radiografía de mano y muñeca izquierdas. Sus resultados deben orientarnos ampliamente en el enfoque del paciente. La realización adicional de estudios moleculares, cuando se sospeche una enfermedad monogénica, y la necesidad de practicar estudios cardiológicos, oftalmológicos, esqueléticos, psicológicos y paidopsiquiátricos, deberá efectuarse cuando proceda a la luz de la información clínica y de los estudios complementarios antes comentados.
En esta revisión se analizarán las bases etiológicas y los fundamentos diagnóstico-terapéuticos de las principales causas de hipercrecimiento.
Somatic overgrowth is a complex and heterogeneous pathology that is only partially understood, although developments in molecular biology have allowed the discovery of the aetiological basis of some of these conditions. The differential diagnosis of a patient with a possible variant of normality, a chromosomopathy, a dysmorphic syndrome, a metabolic or an endocrine disease is essential. The initial clinical evaluation should include a correct anamnesis and physical examination, as well as complementary laboratory and image analyses that will help to orient the diagnosis. This should include a full blood counts and complete biochemical analysis, determinations of IGF-I, IGFBP-3, free T4, TSH and homocystinuria, as well as a karyotype and an X-ray of the left hand and wrist. These results should be very beneficial in orienting the diagnosis. Additional molecular studies should be performed when a monogenic disease is suspected. Cardiological, ophthalmological, skeletal, psychological and psychiatric studies should be performed if the clinical information and previously mentioned complementary studies so indicate.
In this review, the aetiological basis and the diagnostic-therapeutic principles in the most common causes of overgrowth, will be analysed.
El crecimiento excesivo es menos frecuente que el hipocrecimiento debido a que diversos factores socioculturales han contribuido a que la talla alta sea considerada como un índice de buena salud. En consecuencia, es excepcional que los padres de un niño acudan a la consulta porque este sea excesivamente alto, a no ser que se trate de un hipercrecimiento o gigantismo dismórfico. Sin embargo, al lado de situaciones que representan variaciones extremas de la normalidad, existen cuadros patológicos en los cuales la talla alta puede ser una manifestación de enfermedades graves o forma parte de síndromes complejos, que asocian crecimiento excesivo, visceromegalia, alteraciones sistémicas y tendencia a padecer tumores malignos. Se estima que en torno al 2,5% de la población tiene una estatura por encima de 2 desviaciones estándar o del 97,5% de la media.
Las manifestaciones del hipercrecimiento pueden variar. En efecto, este puede afectar al cuerpo en su totalidad, influyendo sobre el crecimiento lineal y dando como resultado una talla adulta elevada, o puede afectar solamente a una región (hemihipertrofia, macrocefalia o macrodactilia) o a un sistema (obesidad o maduración esquelética avanzada). Finalmente, en otras ocasiones, el hipercrecimiento puede ser transitorio, cursando con un incremento en la velocidad de crecimiento durante la infancia y con una talla adulta baja, como acontece en los casos de pubertad precoz, ya central, ya periférica.
Los síndromes con hipercrecimiento global, cursan con talla alta, definida por una altura en bipedestación mayor de +2 DE para la media de la misma población y sexo o velocidad de crecimiento excesiva, ya prenatal, ya posnatalmente. El reconocimiento y diagnóstico de los cuadros clínicos que cursan con hipercrecimiento son relevantes para un adecuado tratamiento médico, correcto consejo genético y vigilancia de aparición de posibles procesos tumorales.
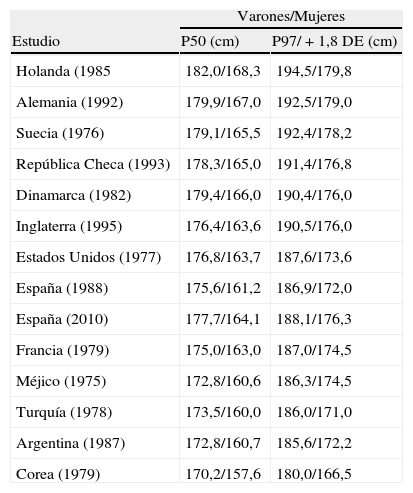
En la tabla 1 se refleja el promedio de estatura de varones y mujeres de diferentes países (P50 y P97).
Promedio de estatura (cm) en varones y mujeres en diferentes países
| Varones/Mujeres | ||
| Estudio | P50 (cm) | P97/+1,8 DE (cm) |
| Holanda (1985 | 182,0/168,3 | 194,5/179,8 |
| Alemania (1992) | 179,9/167,0 | 192,5/179,0 |
| Suecia (1976) | 179,1/165,5 | 192,4/178,2 |
| República Checa (1993) | 178,3/165,0 | 191,4/176,8 |
| Dinamarca (1982) | 179,4/166,0 | 190,4/176,0 |
| Inglaterra (1995) | 176,4/163,6 | 190,5/176,0 |
| Estados Unidos (1977) | 176,8/163,7 | 187,6/173,6 |
| España (1988) | 175,6/161,2 | 186,9/172,0 |
| España (2010) | 177,7/164,1 | 188,1/176,3 |
| Francia (1979) | 175,0/163,0 | 187,0/174,5 |
| Méjico (1975) | 172,8/160,6 | 186,3/174,5 |
| Turquía (1978) | 173,5/160,0 | 186,0/171,0 |
| Argentina (1987) | 172,8/160,7 | 185,6/172,2 |
| Corea (1979) | 170,2/157,6 | 180,0/166,5 |
En la tabla 2 se reflejan las alteraciones que cursan con hipercrecimiento, enumerando los principales factores que regulan el crecimiento (genéticos, nutricionales y hormonales). La mayoría de estas anomalías son primarias, existiendo un número aislado de casos de macrosomía, que son difíciles de clasificar y no se han incluido.
Clasificación de talla alta o hipercrecimiento
| I. HIPERCRECIMIENTO POSTNATAL |
| A. Variantes Normales |
| • Talla alta familiar (genética) |
| • Maduración rápida familiar (genética) |
| B. Nutricional |
| • Obesidad |
| C. Hormonal |
| 1. Exceso de hormona de crecimiento (GH): |
| • Gigantismo hipofisario: |
| • Adenoma hipofisario |
| • Síndrome de McCune-Albright |
| • Adenomatosis endocrina múltiple (MEN-1) |
| • Adenomas ectópicos (cavidad nasal-esfenoidea) |
| • Exceso de GHRH: |
| • Gangliocitomas intracraneales |
| • Tumores extracraneales (carcinoide, pancreático, adenomas bronquiales, etc.) |
| 2. Acromegaloidismo |
| 3. Receptores de Factores de Crecimiento: |
| • Trisomía del IGF1R |
| • Alteración del FGFR3 (S. CATSHL) |
| 4. Hipertiroidismo |
| 5. Hiperinsulinismo: |
| • Neonatos y lactantes |
| • Lipodistrofia |
| 6. Exceso prepuberal de hormonas sexuales: |
| • Pubertad precoz isosexual |
| • Andrógenos o estrógenos suprarrenales |
| • Andrógenos o estrógenos gonadales |
| 7. Deficiencia o insensibilidad de hormonas sexuales: |
| • Eunucoidismo: |
| • Varón–Deficiencia testicular hipogonadotrópica |
| • Mujer - Hipogonadotrópico |
| • Resistencia estrogénica y deficiencia de aromatasa |
| • Resistencia androgénica |
| • Disgenesia gonadal XY (síndrome de Swayer) |
| • Deficiencia de 17-hidroxilasa XY |
| 8. Deficiencia glucocorticoidea familiar (mutación en el gen del receptor de ACTH) |
| D. Genético: |
| 1. Cromosomopatías: |
| • Trisomía X (mujeres 47,XXX) |
| • Klinefelter XXY, XXYY |
| • Varones XYY |
| • Síndrome de sromosoma X frágil |
| • Deleción 22q13.3 |
| 2. Síndromes y otros: |
| • Síndrome de Marfan |
| • Síndrome de Beals (CCA) |
| • Fibrillinopatías |
| • Homocistinuria |
| • Síndrome de Beckwith-Wiedemann |
| • Hipercrecimiento somático (metilación H19) |
| • Síndrome de Simpson-Golabi-Behmel |
| • Síndrome PTEN (Hamartoma) |
| • Síndrome de Partington |
| • Síndrome de Sotos |
| • Síndrome de Weaver |
| • Neurofibromatosis–Tipo I |
| • Síndrome de Nevo |
| • Síndrome de Elejalde |
| II. HIPERCRECIMIENTO PRENATAL |
| • Hijo de madre diabética |
| • Lactante gigante |
| • Síndrome de Beckwith-Wiedemann |
| • Síndrome de Simpson-Golabi-Behmel |
| • Lipodistrofia |
| • Síndrome de Sotos |
| • Síndrome de Weaver |
| • Síndrome de Nevo |
| • Síndrome de Marshall-Smith |
| • Síndrome de Perlman |
| • Síndrome de Elejalde |
Las manifestaciones clínicas de un buen número de síndromes de hipercrecimiento pueden solaparse, haciendo difícil el diagnóstico y requiriendo estudios genéticos, En la evaluación diagnóstica de pacientes con talla alta, puede ser de ayuda el algoritmo que se refleja en la figura 1.
- •
La historia clínica y el examen físico son de gran importancia. El análisis de la curva de crecimiento y el peso, así como las tallas de sus padres, son de gran valor. En cualquier caso, debe analizarse la edad ósea y la predicción de talla adulta.
- •
El estudio bioquímico excluye anomalías metabólicas y disfunción orgánica.
- •
La determinación de los niveles séricos de IGF-I e IGF-II es precisa para descartar cuadros clínicos de hipersecreción de hormona de crecimiento (GH).
- •
Dado que la trisomía X en la mujer (XXX) y el síndrome de varones XYY pueden cursar con talla alta, es preciso efectuar un cariotipo para descartar estos síndromes.
- •
Es necesario determinar los niveles séricos y urinarios de homocisteína ante cualquier paciente con retraso mental, anomalías del sistema nervioso central o elementos marfanoides; asimismo, es imprescindible en niñas con talla alta antes de iniciar tratamiento con estrógenos.
- •
Dada la alta incidencia de retraso mental en el síndrome X frágil, particularmente en varones, el estudio de ADN es necesario.
- •
Otras determinaciones dependerán de la presencia de signos y síntomas como la realización de un ecocardiograma con medición de la raíz de la aorta en pacientes con sospecha de síndrome de Marfan; dosificación de los niveles séricos de gonadotropinas y hormonas sexuales en pacientes con anomalías en el desarrollo puberal; resonancia magnética craneal en pacientes con sospecha de tumores en el área hipotálamo-hipofisaria y estudios moleculares en casos específicos.
- •
Cualquier paciente que presente rasgos dismórficos y otras anomalías (retraso mental) probablemente estará afecto de un síndrome.
- •
Cualquier paciente con retraso marcado o ausencia de pubertad puede tener un hipogonadismo o una disgenesia gonadal XY. Los pacientes con pubertad precoz pueden tener un origen central o periférico.
- •
Ante cualquier paciente obeso sin otros hallazgos físicos anómalos, la «sobrenutrición» suele ser la causa más probable de su talla alta. No obstante, ante pacientes con obesidad mórbida de inicio precoz es necesario practicar estudios moleculares.
- •
Ante cualquier paciente con marcada ausencia de tejido subcutáneo, la causa más probable es la lipodistrofia.
- •
Cualquier paciente con talla alta y buena salud, sin hallazgos físicos anómalos e hijo de padres altos, lo más probable es que sea diagnosticado de «talla alta genética», una presunta variante de la normalidad. No obstante, podría padecer una «deficiencia de aromatasa» o una «resistencia estrogénica», inicio de un «gigantismo hipofisario», una «resistencia androgénica» o una «cromosomopatía». Para efectuar el diagnóstico diferencial, deberá efectuarse un cariotipo, además de valorarse los niveles séricos de IGF-I.
- •
En el caso de pacientes con retraso mental y rasgos dismórficos con cariotipo normal, disponemos en la actualidad de métodos de gran valor para el análisis del genoma completo, como la «hibridación del genoma comparativa de alta resolución» (array-HR-CGH) y el «Multiplex Ligation Probe Amplification» (MLPA) para detectar disbalances cromosómicos (microdeleciones-duplicaciones) y nuevas regiones cromosómicas, así como genes candidatos para fenotipos específicos1,2 y, más recientemente, el exoma.

Datos recientes han detectado diferentes genes y factores involucrados en el crecimiento proporcional y lineal3. Las bases moleculares de los diferentes cuadros de hipercrecimiento (establecidas o sugeridas) quedan reflejadas en la tabla 3 y, pueden resumirse, como sigue:
- I.
Exceso del gen SHOX en el cromosoma X extra en los pacientes con síndrome de Klinefelter y en la trisomía X. El gen SHOX se localiza en la región pseudoautosómica 1 (PAR1) de los cromosomas sexuales (X e Y). Asimismo, un exceso del gen Growth control gene–GCY- (cartografiado en el brazo largo del cromosoma Y), como acontece en varones XYY.
- II.
Exceso de secreción de hormona de crecimiento en el gigantismo hipofisario, síndrome de McCune-Albright (20q12-q13.2), enfermedad neoplásica múltiple tipo I (MEN I -11q13-) y el complejo de Carney tipo II (2p16).
- III.
Exceso o modulación de los factores de crecimiento (IGF-I, IGF-II e insulina):
- A.
Sobreexpresión de IGF-II en el síndrome de Beckwith Wiedemann (BWS) (gen IGF2, 11p15.5) y en el hipercrecimiento somático observado en la metilación anormal y silenciamiento del H19 (11p15.5); o la modulación de IGF-II en el síndrome de Simpson-Golabi-Behmel (SGBS) (alteración en el gen del glipicano 3, Xq26) y SGBS tipo II (Xp22).
- B.
Exceso de insulina (e IGF-I libre) en obesidad, y exceso de insulina en lipodistrofia, lactantes de madres diabéticas y lactantes gigantes con hiperinsulinemia. Entre los síndromes de hiperinsulinemia neonatal, se han descrito recientemente 5 formas genéticas: mutaciones en homocigosis según patrón autonómico recesivo en los genes KCNJ11 y ABCC8 (que codifican para las subunidades del canal KATP) de las células β del páncreas (subunidades Kir6.2 y SUR1, respectivamente),(11p15.1); mutaciones activadoras según patrón autosómico dominante del gen de la glucoquinasa(GCK) [7p15-p13]; hiperinsulinismo hiperamoniémico según patrón autosómico dominante (mutaciones activadoras del gen de la glutamato deshidrogenasa (GLUD1) [10q23.3]) y el gen de la enzima mitocondrial de cadena corta 3-Hidroxi-acil-CoA que cataliza la oxidación de ácidos grasos4. Las anomalías focales en el crecimiento y función de las células β del páncreas debidas a la pérdida del brazo corto materno del cromosoma 11, con la pérdida de los genes ABCC8/KCNJ11 y otros genes supresores, pueden ser causa también de hiperinsulinismo.
- A.
- IV.
Exceso o mutaciones de los receptores de factores de crecimiento: trisomía de IGF1R, en pacientes con trisomía del 15q; mutaciones inactivantes del receptor número 3 del factor de crecimiento de fibroblastos (FGFR3 en 4p16.3.); y en el síndrome de Partington (trisomía de 4p16.3).
- V.
Deficiencia de factores necesarios para detener el crecimiento: estrógenos (deficiencia de aromatasa [15q21.1]), deficiencia del receptor estrogénico (6q25.1) e hipogonadismo.
- VI.
Deficiencia de factores para prevenir la elongación de los huesos y proporciones dismórficas: síndrome de Marfan I (MFS1), por anomalías en el gen de la fibrilina, FBN1 en 15q21.1; síndrome de Marfan II (MFS2), por anomalías en TGFBR2 en 3p22); fibrilinopatías (anomalías de FBN1, en 15q21.1) sin alteraciones clásicas del síndrome de Marfan; síndrome de Beals (mutación en el segundo gen de la fibrilina, FBN2, 5q23-31) y homocistinuria tipo 1 (deficiencia de cistationina β-sintetasa, CBS, 21q22.3).
- VII.
Alteraciones en los genes que afectan al ciclo celular, proliferación, crecimiento y supresión tumoral: como el síndrome PTEN hamartoma (alteraciones del gen PTEN, 10q23.31), que incluye el síndrome de Bannayan-Riley-Ruvalcaba, la enfermedad de Cowden y la enfermedad de Lhermitte-Duclos (todos ellos son síndromes alélicos); síndrome de Sotos (anomalías del gen NSD1, 5q35) y neurofibromatosis tipo 1 (por anomalías en el gen NF1, 17q11.2).
Bases moleculares y etiopatogenia del hipercrecimiento (establecidas o sugeridas)
| Bases moleculares | ||
| Genes de crecimiento extra | ||
| • Klinefelter (47,XXY)• Trisomía X (47,XXX)• Varones 47,XYY | Gen SHOX extraGen SHOX extraGen específico de control del crecimiento Y extra | Extra XExtra XExtra Y |
| Secreción excesiva de GH (tumores hipofisarios) | ||
| • Esporádico:• Gigantismo/Acromegalia• McCune-Albright | Mutaciones en gen Gs αLOH (pérdida alélica)(sin mutaciones del gen MEN1)Sobreexpresión de PTTG | 20q12-q13.211q13 |
| • Familiar:• MEN-1• Acromegalia/Gigantismo | Mutaciones en gen Gs αMutaciones en gen MEN1y LOH de 11q.13LOH (pérdida alélica)(sin mutaciones de MEN-1) | 20q12-q13.211q1311q13 |
| Factores de crecimiento extra | ||
| • IGF-II:• Beckwith-Wiedemann• Silenciamiento de H19• Simpson-Golabi-Behmel, tipo 1• Simpson-Golabi-Behmel, tipo 2• IGF-I-Insulina:• Obesidad• Lipodistrofia• Lactante de madre diabética• Lactantes gigantes con hipoglucemia neonatal | Sobreexpresión de IGF2Sobreexpresión de IGF2Deficiencia de glipicano 3 - GPC3Mutación en CXORF5LEP, LEPR, MC4R, MC3RBSCL2, AGPAT2,CAV1,LMNA, PPARγZMPSTE24, AKT2, CIDECHiperinsulinismoKCNJ11, ABCC8, GCK, GLUD1 | 11p15.511p15.5Xq26Xp227q31.3, 1p31, 18q22, 20q13.211q13, 9q34.3, 7q31.1, 1q21.2, 3p25, 1p34, 19q13.1-q13.2, 3p25.311p15.1, 11p15.1, 7p15-p13, 10q23.3 |
| Factores de crecimiento–receptores | ||
| • Trisomía de IGF-1R• Síndrome CATSHL• Síndrome de Partington | IGF-IR ExtraMutación inactivadora def FGFR3¿Gen FGFR3 extra? | Duplicación de 15q4p16.3Duplicación de 4p16.3 |
| Deficiencia de factores necesarios para detener el crecimiento | ||
| • Deficiencia de aromatasa• Deficiencia de receptor estrogénico• Hipogonadismo | Deficiencia estrogénica - mutaciones–gen Cyp19Deficiencia estrogénica-Mutaciones-receptor estrógenos αDeficiencia estrogénica (secundaria) | 15q21.16q25.1Varias |
| Deficiencia de factores necesarios para prevenir la elongación de los huesos | ||
| • Marfan I (MFS1)• Marfan II (MFS2)• Fibrillinopatías• Beals (CCA)• Homocistinuria tipo 1 | Anomalías de gen de fibrilina (FBN1)Mutaciones en TGFBR2Mutación del gen de fibrilina (FBN1)Mutación en FBN2Mutaciones de CBS | 15q21.13p24.115q21.15q23-3121q23 |
| Alteraciones de genes–relacionados con la regulación del ciclo celular, crecimiento y supresión tumoral | ||
| • Síndrome PTEN hamartoma• Bannayan-Riley-Ruvalcaba• Enfermedad de Cowden• Enfermedad de Lhermitte-Duclos• Síndrome de Sotos• Neurofibromatosis tipo 1 | Mutaciones en PTENMutaciones en PTENMutaciones en PTENMutaciones en PTENMutaciones en NSD1Anomalías en NF1 | 10q2310q2310q2310q235q3517q11.2 |
CBS=cistationina-beta-sintetasa; CCA=aracnodactilia congénita contractural; Cyp19=citocromo P450, 19, aromatasa; FBN1 or FBN2=gen de fibrilina 1 o 2; FGFR-3=receptor 3 del factor de crecimiento de fibroblastos; GPC3=glipicano 3; Gs α=guanine nucleotide-binding protein, 1 stimulatory, alpha chain; IGF-I o -II=factor de crecimiento similar a la insulina I o II; LOH=pérdida de heterocigosidad; MEN1=neoplasia endocrina múltiple tipo 1; NF1=neurofibromatosis tipo1; NSD1=nuclear receptor binding SET domain protein 1; PTEN=fosfatasa y homólogo de tensina; PTTG=pituitary tumor transforming gene; SHOX=short stature homeobox containing gene; TGFBR2=transforming growth factor-beta receptor 2.
Incluye cualquier niño con talla alta, por lo demás normal, que madura adecuadamente y desarrolla su pubertad normalmente en el momento correcto, presentando una talla adulta alta que podriamos denominar «idiopática», pues la etiología aún no ha sido establecida.
La talla adulta es uno de los rasgos humanos con más componente hereditario, creyéndose es poligénica5.
Lettre G et al6 seleccionaron 150 single nucleotide polymorphisms (SNP) de 8 genes candidates en el eje GH/IGF-I (GHR, GHRH, GHRHR, IGF1, IGFALS, IGFBP3, JAK2, STAT5B). Estos autores no observaron ninguna asociación consistente entre la talla y las variantes comunes en estos 8 genes, incluyendo una deleción común del exón 3 del gen GHR. Dichos resultados indican que las variaciones comunes en los genes del eje GH/IGF-I no representan un factor determinante de la talla.
La secreción de GH es variable en los varones altos normales; sin embargo, no se aprecian diferencias significativas en los estudios de secreción de 24 horas de GH e IFG-I en adultos jóvenes de talla normal o talla alta7. Estudios diferentes han sugerido la posibilidad de un grado de sensibilidad variable del GHR7,8.
Garrone et al9 detectaron que las concentraciones de IGF-I, IGFBP-3, y ALS, en niños altos, no son significativamente diferentes de las encontradas en los sujetos control. Por el contrario, las concentraciones de IGF-II e IGFBP-2 son significativamente mayores, mientras que las de IGFBP-1 son menores en niños prepuberales comparados con controles. La proporción molar IGF-I e IGF-II/IGFBPs–1, 2 y 3 es significativamente mayor en niños altos que en controles, especialmente el ratio IGF-II/IGFBP, responsable del exceso del péptido IGF en relación a las concentraciones de IGFBP. Por consiguiente, existe una mayor disponibilidad de IGF libre en los tejidos diana que pudiera ser responsable del hipercrecimiento en los niños con talla alta.
Los polimorfismos en el gen FBN1 del síndrome de Marfan pueden afectar a la talla de sujetos normales. Mamada et al10 genotiparon 3 SNPs exónicos en 219 varones con talla alta y 209 varones con talla normal. Uno de los SNPs (#rs8033037), en el exón 18, mostró una correlación significativa con la talla adulta, sugiriendo que FBN1 es uno de los genes que intervienen en la talla en los individuos normales.
Los sujetos altos normales sin anomalías que sugieran un síndrome específico pueden presentar mutaciones en el gen FBN111, mientras que otros presentan metilación anómala y silenciamiento del gen H19 y sobreexpresión de IGF2. Esto hace que, en ocasiones, pueda ser difícil saber si un sujeto alto es normal sin efectuar un genotipado amplio.
Niñas altas normalesHabitualmente no es difícil establecer el diagnóstico de talla alta en una niña normal, con los datos obtenidos de la historia clínica, el examen físico, el desarrollo puberal normal, la ausencia de rasgos dismórficos y la historia familiar de talla alta.
La talla alta en mujeres es en la actualidad mejor aceptada y, habitualmente, no se considera una limitación12. En consecuencia, la solicitud de tratamiento para disminuir la talla adulta es cada vez menos frecuente. El tratamiento consiste en el empleo de estrógenos a razón de 3 a 10 veces la dosis utilizada como tratamiento de sustitución en caso de deficiencia13–15.
La reducción de la talla depende de la dosis estrogénica y del potencial de crecimiento existente al inicio del tratamiento, pudiendo establecerse entre 2,5cm en una niña con 14 años de edad ósea a 14cm si la edad ósea se sitúa entre 10 1/2 u 11 años14.
Los efectos secundarios graves son infrecuentes, siendo la trombosis la complicación más relevante. En efecto, en un seguimiento de 904 casos, únicamente se demostró un episodio troemboembólico en una niña en tratamiento, tras una herida por aplastamiento del pié. Se han publicado los efectos de la fertilidad a largo plazo tras altas dosis de estrógenos en niñas altas16.
Por consiguiente, dado el riesgo de trombosis y la posibilidad de alteraciones en la fertilidad, no debe promoverse este tratamiento, si bien puede ofrecerse en casos seleccionados y bien justificados, dado el gran beneficio que puede obtenerse en algunas pacientes. Este tratamiento puede aplicarse y ser de ayuda en diferentes pacientes son síndromes de hipercrecimiento (Marfan, Sotos, trisomía X y resistencia androgénica, entre otros); sin embargo, debe contraindicarse en pacientes con homocistinuria, y en todos aquellos casos con predisposición trombótica (por ejemplo: deficiencia de proteína C, proteína S, AT-III, presencia de factor V de Leiden, mutación en el gen de protrombina o deficiencia de plasminógeno).
Niños altos normalesLa talla alta en niños se acepta mejor socialmente, así como por el propio individuo, lo que hace que sea un acto médico excepcional.
Habitualmente el diagnóstico no es complejo, al estar ante un niño alto, con una historia clínica y examen físico normales, desarrollo puberal, asimismo normal y ausencia de rasgos dismórficos, con antecedentes familiares de talla alta. En cualquier caso, deberá valorarse la edad ósea y obtenerse la predicción de talla adulta.
El tratamiento con ésteres de testosterona de acción prolongada puede reducir la talla adulta14. No obstante, es excepcional que estos niños requieran tratamiento o soliciten tratamiento para reducir su talla adulta. De utilizarse, se aplica cuando la predicción de talla adulta es superior a 198cm. Las preparaciones más habitualmente empleadas son el enantato o cipionato de testosterona a razón de 200mg cada 2 semanas para adolescentes jóvenes y 500mg cada 2 semanas en adolescentes mayores.
La reducción de la talla dependerá de la edad ósea al inicio del tratamiento (3,0cm±2,29), en aquellos en los que la edad ósea es superior a 15 años al inicio del tratamiento, a 8,0cm±5,4 en varones con edad ósea de 12 a 14 años. No obstante, estas cifras son variables. Así, en un varón de 12 años con una predicción de talla adulta de 203cm y una edad ósea de 9 ½ años, tratado con enantato de testosterona (200mg cada 2 semanas) durante 1 ½ años, la edad ósea avanzó 6 años (4 años por año de edad cronológica), obteniendo una reducción de talla de 21,5cm.
Este tratamiento puede aplicarse también a pacientes con síndromes de hipercrecimiento, como el síndrome de Klinefelter (XXY), varones XYY, eunucoidismo y síndrome de Marfan.
La bromocriptina disminuye el porcentaje de crecimiento, pero no avanza la edad ósea, por lo que su empleo en reducir la talla adulta no parece aconsejable.
El octreótido tampoco reduce la talla final de forma suficiente para justificar su empleo17.
Una reducción de la talla adulta comparada a su predicción de 7cm (1,2-13,8cm) en pacientes con predicción de talla adulta excesiva puede conseguirse mediante epifisiodesis percutánea bilateral en la rodilla. Este procedimiento normaliza las proporciones corporales18.
Nutrición–Sobrenutrición (obesidad)Los niños obesos tienden a incrementar su velocidad de crecimiento, a ser más altos que los niños delgados de su edad y a presentar una maduración esquelética más avanzada. Las niñas presentan pubertad y menarquia precoces. El porcentaje de maduración sexual en varones es variable. Aunque los niños prepuberales obesos sean más altos que los delgados de su edad, no alcanzan una talla adulta alta o excesiva.
Se han descrito anomalías monogénicas que cursan con obesidad, debidas a mutaciones en los genes de: leptina, receptor de leptina, POMC, prohormona convertasa 1, factor neurotrófico derivado del cerebro (BDNF), receptor quinasa tirosina (Trkβ) y, las más frecuentes entre ellas, por afectación del receptor 4 de melanocortina (MC4R)19, entre otros.
La talla alta y la velocidad de crecimiento acelerada en estos pacientes está probablemente mediada por la insulina y el IGF-I. En efecto, los niños obesos presentan niveles elevados de IGF-I, a pesar de tener suprimidos sus niveles de GH, comparado a los niños de peso normal y de su misma edad. La hiperinsulinemia es frecuente en pacientes obesos, cursando con niveles séricos disminuidos de IGFBP-1 y elevados de IGF-I libre20,21. Estos cambios hormonales pueden revertirse, al menos parcialmente, con la pérdida ponderal.
El incremento de la velocidad de crecimiento en los niños obesos es probablemente debido a la hiperinsulinemia y al aumento de los niveles de IGF-I libre.
HormonalSecreción excesiva de GHGigantismo hipofisario y acromegaliaEl gigantismo se caracteriza por talla alta y alargamiento desproporcionado de las partes acras, manos y pies, acompañado de factores faciales anómalos (alargamiento de arcos supraorbitarios, nariz, pabellones auriculares, labios y pliegues nasolabiales) (fig. 2).

Tanto el gigantismo como la acromegalia pueden deberse a una secreción excesiva de GH, por la existencia de una hiperplasia o un adenoma eosinofílico o cromófobo de la adenohipófisis, la existencia de secreción ectópica de GHRH central por tumores hipotalámicos (gangliocitomas) o periférica (adenomas pancreáticos y tumores carcinoides y bronquiales)22,23. Además, en ciertos adenomas y en pacientes con síndrome de McCune-Albright, se detectan mutaciones en el gen GNAS (OMIM 139320 y OMIM 174800)24. En pacientes con complejo de Carney, se aprecian mutaciones en PRKAR1A (OMIM 188830). Finalmente, los pacientes con neoplasia endocrina múltiple (MEN1) (OMIM 131100) presentan mutaciones en el gen MEN1.
El diagnóstico se basa en la demostración de una secreción excesiva de GH, así como en la presencia de un adenoma hipofisario o hiperplasia hipofisaria con alargamiento selar23,25. El síndrome de McCuneAlbright conforma, aproximadamente, el 20% de los pacientes publicados con gigantismo. El test definitivo para el diagnóstico de secreción de GH excesiva radica en el fallo de la GH para disminuir a menos de 1ng/ml tras un test de tolerancia oral de glucosa (1,75g/Kg–máximo 75g-). Los individuos normales disminuyen la concentración sérica de GH a <1ng/ml. La concentración sérica de IGF-I es un test de screening sensible, encontrándose elevada de 4-10 veces el nivel normal.
La cirugía transesfenoidal es el tratamiento de elección. Si la secreción de GH no se normaliza, puede recurrirse a la radiación hipofisaria y al tratamiento médico con octreótido (un análogo de somatostatina de acción prolongada [SMS201-995]) o bromocriptina, o ambos. El octreótido, además, puede emplearse preoperatoriamente para disminuir el tamaño del adenoma26.
Un nuevo tratamiento está basado en el empleo de «pegvisomant», antagonista del receptor de GH. Este normaliza los niveles séricos de IGF-I en más del 90% de los pacientes, representando el tratamiento médico más eficaz de la acromegalia, aunque la seguridad a largo plazo, en especial en relación con el crecimiento del tumor hipofisario y la toxicidad hepática, aún deban determinarse.
AcromegaloidismoGrupo heterogéneo de alteraciones que afectan a niños y adultos y que se caracteriza por talla alta, crecimiento excesivo y elementos de acromegalia, sin secreción elevada de GH ni IGF-I y sin adenoma hipofisario o hiperplasia.
Las manifestaciones clínicas se asemejan a las que presentan los pacientes con gigantismo y acromegalia con secreción excesiva de GH: talla alta, crecimiento rápido en niños, alargamiento de partes acras, facies acromegaloide, cefaleas, astenia, hiperhidrosis, artralgias e hipertensión en más de la mitad de los casos. Manifestaciones clínicas menos frecuentes son: hipertricosis, parestesia, piel grasienta y maloliente y disfonía.
Aunque infrecuente, se han descrito niños sin exceso de GH con acromegaloidismo27,28.
Receptores de factores de crecimientoTrisomía del receptor de IGF-IDescrita en un número pequeño de niños, se asocia frecuentemente con talla alta y retraso mental. En 2002, Faivre et al29 publicaron 4 niños de 2 familias no relacionadas, que cursaban con hipercrecimiento y una duplicación terminal del brazo largo del cromosoma 15. En ambos casos el análisis cromosómico de sus padres mostró una translocación balanceada de 15q26.1-qter. Los estudios moleculares y citogenéticos mostraron 3 copias del gen IGF1R, sugiriendo que este síndrome de hipercrecimiento pudiera estar relacionado con un efecto de dosis del gen IGF1R, en contraposición a lo que acontece en pacientes con retraso del crecimiento severo con deleción de 15q. Se han descrito, con posterioridad, diferentes casos30–32.
Los fibroblastos de un paciente con 3 copias del IGF1R mostraron crecimiento acelerado, mientras que las células de un paciente con una única copia de IGF1R mostró crecimiento enlentecido. El niño con 3 copias del IGF1R fue alto.
Estos hallazgos son consistentes con el concepto de que el número de copias del gen IGF1R es de importancia clínica y funcional en el ser humano, al tiempo que responsable del hipercrecimiento31
Alteraciones del FGFR3–Síndrome de CATSHLEl receptor 3 del factor de crecimiento fibroblástico (FGFR3) es un regulador negativo del crecimiento óseo encondral. Las mutaciones activadoras de FGFR3 causan una variedad de displasias óseas y síndromes de craneosinostosis, incluyendo: acondroplasia, hipocondroplasia, displasia tanatofórica tipo I y tipo II, síndrome de acondroplasia severa con retraso del desarrollo y acantosis nigricans (SADDAM) y síndrome lácrimo-aurículo-dental-digital (LADD).
Recientemente, Toydemir et al33 descubrieron una mutación nueva inactivadora del gen FGFR3, causante de un síndrome caracterizado por camptodactilia (90%), talla alta (100%), escoliosis y pérdida auditiva (85%) consistente en sordera neurosensorial bilateral congénita desarrollada precozmente en la infancia y progresiva, siendo variable (de moderada a severa), conformando el síndrome de CATSHL. Los autores investigaron un amplio árbol genealógico en el que estudiaron 27 miembros familiares afectados vivos, correspondientes a 4 generaciones, evidenciándose un patrón de herencia dominante. Los adultos varones presentaban una media de 195cm de talla y, las mujeres, de 178cm. Algunos de ellos presentaban microcefalia y, en torno al 60% mostraban retraso puberal y/o retraso mental. Los hallazgos radiológicos evidenciaron cuerpos vertebrales altos de bordes irregulares, metáfisis femorales anchas y alargamiento tubular.
Se identificó una mutación sin sentido en heterocigosis del FGFR3 causando una pérdida parcial de FGFR3 en 20 de los 20 miembros afectados que fueron analizados. Esta observación sugiere que la haploinsuficiencia produjo pérdida de la función de FGFR3 por un mecanismo dominante negativo33.
Las anomalías observadas en estos miembros de la familia identificaban plenamente las alteraciones apreciadas en ratones con ausencia de fgfr3 (fgfr3-/-). En efecto, estos ratones muestran huesos largos, en especial el fémur, y cuerpos vertebrales alargados que predisoponen a la cifoescoliosis torácica. Téngase en consideración que, tanto en estos ratones como en humanos con este síndrome, únicamente se encontraban afectados los huesos formados por osificación endocondral. El ratón muestra una profunda sordera neurosensorial, causada por anomalías cocleares.
HipertiroidismoLa talla alta se ha observado en muchos niños con hipertiroidismo. Se desconoce aún si las hormonas tiroideas tienen un efecto sinérgico sobre IGF-I para estimular el crecimiento. Dicho crecimiento se normaliza con el tratamiento del hipertiroidismo.
LipodistrofiasIncluye un grupo infrecuente de enfermedades caracterizadas por ausencia generalizada o parcial de los depósitos de tejido adiposo, resistencia a la insulina, hiperinsulinemia, hiperlipidemia y diabetes mellitus no cetósica.
Los síndromes lipodistróficos pueden ser de origen genético o adquirido. Se han descrito mutaciones en 8 genes causantes de lipodistrofia (BSCL1, BSCL2, AGPAT2, CAV1, LMNA, PPARγ, ZEMPSTE24 y AKT2).
Estudios recientes en relación con las bases genéticas de las lipodistrofias han demostrado que la existencia de pérdida bialélica de mutaciones en BSCL2 (11q13), AGPAT2 (9q34.3) y CAV1 (7q31.1) acontece en más del 90% de todos los casos de lipodistrofia congénita generalizada, mientras que la presencia de mutaciones en heterocigosis en LMNA (1q21.2) y PPARγ (3p25) están presentes en más del 50% de todos los casos heredados de lipodistrofia parcial. Asimismo, se han descrito mutaciones raras en homocigosis y en heterocigosis compuesta en LMNA y en ZMPSTE24 (1p34) y, en una familia con lipodistrofia parcial, una mutación en heterocigosis en AKT2 (19q13.1-q13.2).
Recientemente, hemos publicado una nueva causa de lipodistrofia parcial familiar, con fenotipo similar a las causadas por mutaciones en LMNA, PPARG y AKT2, con lipodistrofia fémorogluteal, esteatosis hepática, dislipidemia y resistencia a la insulina, debida a la primera mutación descrita en el gen CIDEC (3p25.3) –mutación sin sentido en homocigosis-34.
Observaciones recientes en ratones transgénicos y en humanos han demostrado que la resistencia a la insulina, la hiperinsulinemia y la diabetes mellitus son consecuencia de la ausencia de grasa resultante de la deficiencia de leptina, adiponectina y otras adipoquinas. La leptina desempeña un papel relevante en la ingesta de alimentos, gasto energético y funciones neuroendocrinas.
Los pacientes con lipodistrofia generalizada congénita autosómica recesiva (BSCL) presentan gigantismo, velocidad de crecimiento acelerada, elementos acromegaloides, manos, pies y orejas grandes, mandíbula prominente y edad ósea acelerada. Pueden presentar macrosomía al nacimiento o gigantismo posnatal. La hiperinsulinemia puede ser responsable de la hipertrofia muscular, síndrome de ovario poliquístico e incremento del crecimiento. La secreción de GH es normal o baja; sin embargo, la insulina puede activar el receptor de IGF-I, generando el crecimiento acelerado, así como los cambios acromegaloides.
En ausencia de depósitos de tejido adiposo, el exceso de lípidos se acumula en tejidos no-adiposos, como hepatocitos, cardiomiocitos, células esqueléticas o células β. Estas células almacenarán la grasa para su empleo como energía en momentos de necesidad. La esteatosis hepática y los triglicéridos miocelulares se asocian con resistencia a la insulina debido a alteración en la oxidación de los ácidos grasos.
Todos los pacientes con lipodistrofia presentan niveles séricos bajos de leptina y de adiponectina, asi como grados variables de resistencia a la insulina34. Yamauchi et al35 han demostrado que la resistencia a la insulina en ratones con lipodistrofia generalizada revierte con la administración sistémica de adiponectina y leptina, y solo parcialmente con la administración individualizada de adiponectina o de leptina. Del mismo modo, Oral et al36 han demostrado en el ser humano una gran mejoría en el control glucémico, de la hipertrigliceridemia y de la esteatosis hepática en pacientes tratados con leptina recombinante (0,04 a 0,08mg/Kg/día) durante 4 meses. No obstante, la mejoría no fue completa. Es posible que, como en el ratón, deba asociarse adiponectina a la leptina para obtener tal vez una mejoría completa.
Exceso prepuberal de hormonas sexualesLa secreción prepuberal de andrógenos o estrógenos, con independencia de su causa, es probablemente la razón más común de velocidad de crecimiento excesiva y de talla alta en la infancia. La pubertad precoz (completa o incompleta, isosexual) cursa con un incremento de la secreción de andrógenos de las glándulas suprarrenales (hiperplasia suprarenal congénita, hiperplasia suprarenal congénita no clásica, resistencia periférica a glucocorticoides, tumores) o testicular (tumores de las células de Leydig) o un incremento en la secreción de estrógenos desde las glándulas suprarrenales o los ovarios (quistes o tumores).
Aunque la talla alta está presente en la infancia, como consecuencia de la edad ósea avanzada, la talla adulta suele ser inferior a la normal si el proceso patológico no es tratado adecuadamente.
Las manifestaciones clínicas de este grupo de enfermedades son distintas37.
Deficiencia o resistencia de hormonas sexualesDeficiencia permanente de testosterona en el varón y de estrógenos en la mujerGenera retraso en la maduración esquelética, crecimiento prolongado en el tiempo, talla alta y proporciones eunucoides, con piernas alargadas y disminución del ratio segmento superior-inferior. Naturalmente, excluyendo los cuadros clínicos de deficiencia de GH, síndrome de Turner u otra enfermedad que afecte el desarrollo del crecimiento normal.
La deficiencia de testosterona en el varón puede ser el resultado de la existencia de deficiencia en gonadotrofinas de diferentes orígenes, alteración en su propia producción o anomalías enzimáticas en la síntesis de testosterona. La deficiencia de céulas de Leydig o la anorquia son, asimismo, causas de deficiencia de testosterona.
La deficiencia estrogénica en la mujer puede ser el resultado de la existencia de deficiencia de gonadotrofinas, alteración en su propia producción o anomalías enzimáticas en la síntesis de estrógenos (ejemplo: deficiencia de 17-alfa-hidroxilasa, deficiencia de aromatasa), o ausencia de ovarios.
En varones con deficiencia de testosterona, para prevenir la ausencia de desarrollo puberal, anomalías psicológicas y proporciones eunucoides, debe iniciarse tratamiento con testosterona después de que el paciente alcance una edad ósea de 11 a 12 años. Se prefiere el empleo de inyectables de acción prolongada de ésteres de testosterora (enantato o cipionato). Pueden emplearse diferentes pautas, si bien lo importante consiste en alcanzar un desarrollo puberal completo en un periodo de 4-5 años, a la misma edad que sus compañeros, alcanzando su potencial genético de talla, sin talla alta ni proporciones eunucoides.
Las formas transdérmicas de testosterona, ya sean parches, ya geles, están ya disponibles. Aún se precisa de una mayor información sobre las concentraciones de testosterona obtenidas con 2,5mg de parche o gel en adolescentes jóvenes, así como el efecto sobre la maduración y los cambios puberales antes de recomendarlos.
En mujeres con deficiencia estrogénica, el tratamiento con estrógenos debe iniciarse cuando alcancen una edad ósea de 10 a 11 años. Inicialmente debe administrarse una dosis baja de etinil-estradiol (100ng/Kg/día). Como en el caso de los varones, el fundamento radica en que el desarrollo puberal completo se adquiera en un periodo de 4 a 5 años, alcanzando su talla genética y previniendo las proporciones eunucoides.
Deficiencia de aromatasa-Resistencia estrogénicaRecientemente se han descrito algunos varones y mujeres con deficiencia estrogénica debida a deficiencia de aromatasa por mutaciones en el gen CYP19 o resistencia estrogénica, como consecuencia de mutaciones en el gen del receptor de estrógenos α (ERα).
La deficiencia de aromatasa es rara en el ser humano. Dicha enzima cataliza la conversión de andrógenos a estrógenos. Los sujetos afectos, en consecuencia, no pueden sintetizar estrógenos. Si el feto carece de actividad aromatasa, el sulfato de dehidroepiandrosterona producido en sus glándulas suprarrenales no puede convertirse a estrógenos en la placenta, convirtiéndose en testosterona periféricamente y generando virilización del feto y de su madre. Esta virilización se manifiesta como un pseudohermafroditismo en la niña e hirsutismo y acné en la madre.
Hasta la fecha, únicamente se han descrito 7 varones y 7 mujeres con deficiencia de aromatasa.
Las niñas afectas se diagnostican al nacimiento al presentar pseudohermafroditismo, pudiendo presentar quistes ováricos y retraso en la maduración ósea durante la infancia y la adolescencia. En el momento de la pubertad, presentan talla alta, amenorrea primaria, ausencia de desarrollo mamario, virilización e hipogonadismo hipergonadotrófico38.
Por el contrario, los varones afectos no muestran datos claros al nacimiento, por lo que son diagnosticados con posterioridad. Se caracterizan por presentar: talla alta, retraso en la maduración esquelética, retraso en el cierre epifisario, dolor óseo, proporciones corporales eunucoides y exceso de adiposidad, al tiempo que presentan alteraciones en el metabolismo lipídico e insulínico.
Morishima et al39 publicaron un varón de 24 años que medía 204cm y continuaba creciendo, mostrando una edad ósea de 14 años, a pesar de exhibir una pubertad normal y un grado completo de virilización a dicha edad. La densidad mineral ósea se encontraba marcadamente disminuida.
La investigación de este paciente demostró que presentaba una deficiencia severa de aromatasa, debida a una nueva mutación en homocigosis del gen CYP 19 que codifica la aromatasa P-450. Los niveles plasmáticos de andrógenos y gonadotrofinas estaban elevados, mientras que los niveles de estrógenos eran indetectables.
El tratamiento con estrógenos resuelve los síntomas en varones y mujeres40.
Como consecuencia de la ausencia de estrógenos o del efecto estrogénico, el cierre de los cartílagos epifisarios se retrasa extraordinariamente y el periodo de crecimiento se prolonga alcanzando los pacientes una talla alta excesiva.
Smith et al41 publicaron el primer caso de resistencia estrogénica en un varón de 28 años que medía 204cm, que había efectuado un normal desarrollo puberal y se afeitaba regularmente a la edad de 17-18 años. Las concentraciones séricas de testosterona eran normales y el tamaño de sus genitales y su espermatogénesis correspondían a un adulto normal; sin embargo, las epífisis no estaban cerradas y su edad ósea se situaba en torno a 15 años. Finalmente, su densidad mineral ósea se encontraba marcadamente disminuida. Se demostró que presentaba una mutación en el gen ERα responsable de la resistencia estrogénica. Las concentraciones plasmáticas de estrógenos y gonadotropinas se encontraban elevadas.
Este caso clínico, así como los debidos a deficiencia de aromatasa, han demostrado con claridad la función crítica y relevante de los estrógenos en la maduración y cierre epifisario, el estirón de crecimiento puberal, la regulación de gonadotropinas y la mineralización ósea.
Otros cuadros clínicosPuede ser necesario prevenir la inusual talla alta en pacientes con resistencia androgénica completa o incompleta (mujeres XY) y disgenesia gonadal XY (síndrome de Swyer). El tratamiento preventivo de talla alta, si se desea, será similar al empleado en las niñas altas normales.
Los pacientes con deficiencia de 17-alfa-hidroxilasa son genéticamente varones (XY) y podrían ser fenotípicamente mujeres, pudiendo no presentar secreción de andrógenos ni estrógenos y, en consecuencia, ausencia de cambios puberales. La edad ósea se encuentra retrasada y el periodo de crecimiento se prolonga. Estos pacientes necesitan estrógenos para proporcionar los cambios puberales y prevenir su talla alta.
Deficiencia glucocorticoidea familiar (DGF)Se trata de una enfermedad infrecuente que se hereda con un patrón mendeliano autosómico recesivo de falta de respuesta de las glándulas suprarrenales a la ACTH. La talla alta y la edad ósea avanzada son hallazgos frecuentes. Esta enfermedad cursa con deficiencia glucocorticoidea en presencia de niveles séricos elevados de ACTH, pero con producción normal de mineralocorticoides, excepto ocasionalmente en situaciones de estrés42. Habitualmente presentan en la lactancia y en la infancia hiperpigmentación, con electrolitos normales en suero, episodios de hipoglucemia, vómitos y fallo de medro. La edad de comienzo de los síntomas y la severidad clínica de la enfermedad puede variar entre los casos, sugiriendo que se trata de una enfermedad heterogénea de origen genético. Entre los 50 casos descritos, 18 fallecieron como consecuencia de la enfermedad.
Clark et al43 publicaron una mutación puntual en la región codificante del gen del receptor de melanocortina 2 (MC2R) (cromosoma18p11.2) en una familia con esta enfermedad. Algunos casos, no obstante, son homocigotos para la misma mutación y, otros, heterocigotos44.
Entre el 25 al 40% de los pacientes con esta enfermedad presentan una mutación con pérdida de función del gen MC2R, que codifica el receptor de la proteína acopladora ACTH-G (DGF tipo 1–OMIM 202200).
También se han encontrado mutaciones con pérdida de función en un gen (cromosoma 21q22.1) que codifica la proteína accesoria MC2R (MRAP)45 (DGF tipo 2). MRAP ayuda en el proceso de «movilidad» del receptor de ACTH desde el retículo endoplásmico a las membranas plasmáticas de las células productoras de glucocorticoides en la zona fasciculada del córtex suprarrenal.
Un tercer cromosoma (8q11.2-q13.2) se ha detectado en otros sujetos con esta enfermedad, habiéndose denominado DGF tipo 3 (OMIM 609197). El gen mutado aún no ha sido identificado, si bien probablemente codifica un producto en relación con la función del receptor de ACTH.
Los niveles séricos basales de cortisol pueden ser normales en algunos pacientes y, bajos, en otros; sin embargo, la respuesta a la estimulación con ACTH es anormalmente baja.
Los pacientes con mutaciones en gen del receptor de ACTH habitualmente presentan talla alta. El peso y la longitud al nacimiento se sitúan entre los percentiles 75 y 97 y la talla durante la lactancia e infancia precoz se detecta entre +4 y +5,6 DE. No existe ninguna evidencia de secreción excesiva de GH y, los niveles séricos de IGF-I e IGFBP-1 son normales. La edad ósea se encuentra acelerada.
Por consiguiente, la causa de la talla alta no está suficientemente aclarada.
CromosomopatíasGenes extra de crecimientoTrisomía X (mujeres 47,XXX)Las pacientes con trisomía X tienden a ser altas, situándose en torno al percentil 90. Antes de la pubertad las piernas ya son alargadas. La mayoría de las pacientes, sin embargo, muestran un fenotipo normal, por lo que esta cromosomopatía numérica no se diagnostica de no pensar en ella y solicitar un cariotipo. Se trata de la anomalía del X más frecuente, estimándose en 1 de cada 1.000 recién nacidas. Algunas pacientes pueden presentar mínimos rasgos dismórficos (clinodactilia, sindactilia). El desarrollo sexual es normal, así como la menarquia y la función ovárica, si bien algunas pueden presentar amenorrea secundaria e insuficiencia ovárica prematura y, excepcionalmente, se ha descrito amenorrea primaria y disgenesia ovárica. El nivel intelectual se sitúa entre 55 y 115 y, aproximadamente, la mitad a 2/3 de ellas pueden situarse ligeramente por debajo del límite normal46.
El cromosoma X extra probablemente justifica la talla alta. Rao et al en 199747 aislaron la región pseudoautosómica PAR1 en los cromosomas sexuales X e Y, cartografiando el gen que denominaron «short stature homeobox containing gene» (SHOX). Existen evidencias que sugieren que las deleciones de Xp con ausencia de SHOX podrían generar talla baja y la presencia de cromosomas X adicionales con genes SHOX activos (no inactivados) podrían causar talla alta, como ocurre en la trisomía X y en el síndrome de Klinefelter48.
La presencia de extremidades inferiores alargadas en ambas cromosomopatías numéricas antes de la pubertad sugiere que este fenómeno se encuentra relacionado con un cromosoma X extra y que el gen SHOX extra actuaría sobre el crecimiento óseo.
Las mutaciones del gen SHOX producen haploinsuficiencia y se han identificado en aproximadamente el 60% de pacientes con discondrosteosis (síndrome de Léri-Weill), una displasia mesomélica que se hereda de forma autosómica dominante y que cursa con deformidad de Madelung, cambios metafisarios, talla baja y brazos y piernas cortos. Recientemente, hemos identificado una nueva clase de mutaciones en la región PAR1, «aguas abajo» del gen SHOX, en aproximadamente el 15% de estos pacientes con síndrome de Léri-Weill49. Finalmente, la haploinsuficiencia del gen SHOX también se ha encontrado en pacientes con displasia de Langer y en algunos sujetos afectos de talla baja idiopática.
La mayoría de las pacientes con trisomía X no necesitan tratamiento alguno. En caso de talla alta, puede emplearse el mismo tratamiento que el descrito para las niñas altas normales.
Síndrome de Klinefelter XXYDescrito por Klinefelter, Reifenstein y Albright en 1942 con las características clínicas que son evidentes en la adolescencia: ginecomastia, talla alta, grado variable de eunucoidismo, virilización imperfecta, test pequeños de consistencia dura con hialinización de los túbulos seminíferos y agregación de las células de Leydig, pene pequeño e incremento de la excreción urinaria de gonadotropinas (fig. 3). Existen diferentes variantes cromosómicas, entendiéndose por síndrome de Klinefelter en la actualidad, cualquier dotación gonosómica con al menos 2 cromosomas X y un Y, excepto para los escasos pacientes varones XX (46,XX que son SRY positivos).

La talla alta está habitualmente presente antes de la pubertad, siendo la desproporcionada longitud de las piernas lo que sugiere que no está relacionada con deficiencia androgénica y que pudiera estar relacionada con el cromosoma X extra (extra SHOX), como en la trisomía X. La braza no está incrementada. Estos pacientes pueden ser ansiosos, agresivos y cometer conductas antisociales y crueldad hacia los animales. La escala completa de niveles intelectual suele ser normal.
Los pacientes con síndrome de Klinefelter con un isocromosoma Xq (47,XiXqY) carecen del brazo corto extra del segundo cromosoma X y, en consecuencia, del gen SHOX extra, mostrando las características clínicas del síndrome de Klinefelter, excepto la talla alta. Además, los pacientes 46,XX (SRY positivos) con dos genes SHOX, pero sin el brazo largo del cromosoma Y, no son altos, presentando una talla similar a la de las mujeres50.
Las investigaciones cromosómicas en el momento del nacimiento muestran una incidencia aproximada de 1:500 a 1:1.000. Menos del 10% de los fetos estimados se detectan perinatalmente, mientras que el 75% de los pacientes permanecen sin diagnosticar a lo largo de su vida. El diagnóstico de este síndrome después de la pubertad no es difícil, teniendo en cuenta la existencia de: fenotipo típico de talla alta, virilización incompleta, testes pequeños de consistencia dura y niveles séricos elevados de gonadotrofinas51. El diagnóstico debe ser confirmado siempre con el estudio del cariotipo.
Con el inicio de la pubertad, los niveles de testosterona están disminuidos y las gonadotropinas elevadas52, aconteciendo la degeneración de los túbulos seminíferos52,53. La osteoporosis está presente en el 25% de los pacientes. Para prevenir las complicaciones físicas y psicológicas del hipogonadismo, se recomienda el tratamiento con testosterona.
Las alteraciones psicológicas de la ginecomastia persistente requieren mamoplastia reductora. Es necesaria la intervención temprana para el aprendizaje y el tratamiento de las alteraciones de conducta.
Varones 47,XYYLos varones 47,XYY presentan talla alta54. En efecto, el único elemento físico consistente en estos pacientes es la talla alta, encontrándose el 50% por encima del percentil 90.
Se estima que del 40 al 50% presentan dificultades de aprendizaje54. Los adultos con este síndrome son altos, tienen acné quístico nodular, algunas anomalías neurológicas, como temblor intencional, descoordinación y, frecuentemente, sinostosis radio-cubital.
No existen evidencias de endocrinopatías. Los varones XYY son más altos que los varones XY; las mujeres XY son más altas que las mujeres XX, y los pacientes con disgenesia gonadal XY son más altos que los pacientes con disgenesia gonadal XX.
Una región con gran influencia en el crecimiento y en el tamaño de los dientes se localiza en la porción más proximal del brazo largo del cromosoma Y, cercana al centrómero, y es conocida como «Growth Control in the Y» (GCY)55. Aún no se han identificado posibles genes candidatos56. El gen GCY puede ser el responsable de la diferencia en talla y en tamaño dental entre varones y mujeres. Es de interés el estudio longitudinal efectuado en ocho varones XYY no seleccionados. Todos ellos eran más altos que los controles en la infancia y al inicio de la pubertad. La talla adulta fue de 188,1cm. Las observaciones sugieren que la talla alta en varones 47, XYY está en relación con genes de crecimiento extra en el cromosoma Y, SHOX y GCY.
Como en la trisomía X y en el síndrome de Klinefelter, los varones XYY suelen no ser diagnosticados debido a la ausencia de cambios fenotípicos marcados. Para diagnosticarlos es preciso efectuar un cariotipo ante cualquier paciente que presente talla alta de causa desconocida. Los descendientes de estos pacientes tienen un mayor riesgo de padecer cromosomopatías, por lo que el diagnóstico genético preimplantación puede sugerirse a estos pacientes.
Cromosoma X frágilConstituye la forma más común de retraso mental heredado, con una prevalencia estimada de 1:1.250 varones y de 1:2.500 mujeres. En 1991, se aisló y caracterizó el gen responsable, localizado en Xq27.3. El segmento de ADN muestra una peculiar repetición de trinucleótidos (citidina, guanosina, guanosina) en el gen 1 del síndrome X frágil (FMR1) que incrementa el tamaño del fragmento de ADN específico del cromosoma X en Xq27.3. Este síndrome puede acontecer también en sujetos con deleciones y mutaciones puntuales del gen FMR1sin amplificación CGG57,58.
En niñas con síndrome de cromosoma X frágil, los dos hallazgos más importantes son: talla alta (presente desde el nacimiento) y las alteraciones de conducta (incluyendo dificultad de atención severa y timidez y ansiedad extremas). En varones, los datos más tipicos son: retraso mental, talla alta, grandes pabellones auriculares (en el 50% de los casos la longitud de la oreja se sitúa por encima del percentil 90) y macroorquidismo (el volumen testicular después de la pubertad en varones normales se sitúa entre 12 y 25mL y, en estos pacientes, entre 25 y 70mL).
La función intelectual en mujeres oscila entre el límite inferior y el rango medio, mostrando niveles intelectuales inferiores a 70 en el 25% de las pacientes, siendo inferior a 85 en el 53% de las mismas.
El síndrome X frágil asociado a temblor/ataxia (FXTAS) es una enfermedad neurodegenerativa de comienzo tardío que afecta preferentemente al varón y, excepcionalmente, a mujeres portadoras de una premutación (repeticiones de CGG de 55 a 200) en el gen FMR1.
Las mujeres presentan una menor reducción del volumen del cerebelo y una menor incidencia de afectación de los pedúnculos cerebelosos medios (13% comparado con los varones afectos en los que es del 58%)59. La causa de la talla alta aún se ignora.
El síndrome del cromosoma X frágil debe sospecharse tanto en varones como en mujeres con retraso mental y con cambios fenotípicos característicos, ataxia de comienzo tardío, temblor de acción o neuropatía; en especial, en aquellos con historia familiar de retraso mental. Su confirmación requiere estudios de ADN y citogenéticos. No existe ningún tratamiento específico, excepción hecha de las medidas de apoyo psicológico y actuación ante las alteraciones de conducta. Habitualmente no se requiere ningún tratamiento para su talla alta.
Síndrome de deleción del cromosoma 22q13.3 (OMIM #606232)Se han descrito diversos pacientes con deleción terminal del 22q13.3. Dicho síndrome se caracteriza por presentar hipotonía neonatal, retraso global del desarrollo, crecimiento normal o acelerado, retraso en el lenguaje (de ausente a severo), conducta autista y elementos dismórficos menores.
Aunque los pacientes con deleción terminal simple de 22q13 tienen tendencia general a presentar talla alta, los pacientes con cromosoma 22 en anillo padecen a menudo talla baja.
Aún se desconoce la causa de la talla alta en estos pacientes.
Síndromes y otrosSíndrome de Marfan–tipos 1 y 2 (MFS1, MFS2)Se trata de una anomalía heredada del tejido conectivo que afecta al esqueleto, sistema cardiovascular, sistema ocular y ectasia dural del canal espinal lumbosacro. Se estima afecta de 2 a 3 por cada 10.000 personas.
Las manifestaciones clínicas relacionadas con el esqueleto, incluyen: talla alta, brazos y piernas largos y delgados (dolicostenomelia), aracnodactilia («dedos en arena»), pectus excavatum o carinatum, facies estrecha con paladar estrecho y escoliosis y cifosis en 60 al 100% de los casos (fig. 4). La laxitud articular, las hernias inguinales, femorales y diafragmáticas son otras consecuencias del tejido conectivo anormal.
 Adolescente de 16 años y 6 meses de edad con síndrome de Marfan. Detalle de la aracnodactilia de los dedos de manos y pies. Nótese el pectum excavatum B) Varón de 17 años y 4 meses de edad con síndrome de Marfan. Nótese el pectum carinatum.")
Las manifestaciones oculares incluyen: luxación hacia arriba del cristalino, como consecuencia de una anomalía en el ligamento suspensorio, aumento de la longitud axial del globo ocular, con miopía y desprendimiento de retina.
La ectasia dural del canal espinal lumbosacro conforma una de las alteraciones clínicas más importantes.
Las complicaciones más frecuentes que requieren tratamiento de por vida son las que afectan al sistema cardiovascular, con dilatación de la aorta ascendente, con/sin aneurisma disecante y, menos comúnmente, de las arteria aorta torácica o abdominal o de la arteria pulmonar. Como consecuencia de la dilatación de la aorta se produce una regurgitación aórtica secundaria. El prolapso de la válvula mitral es muy frecuente. Sin tratamiento para las complicaciones cardiovasculares, en particular de la dilatación aórtica, la edad media de muerte se sitúa en torno a los 45 años, por disección aórtica y ruptura subsiguiente46.
Son mentalmente normales, pero con alteraciones neuropsicológicas que incluyen: dificultad de aprendizaje y déficit de atención en aproximadamente el 40% de los pacientes.
Se transmite de forma autosómica dominante. Aproximadamente el 85% de los pacientes tienen historia familiar positiva y, en torno a un 15%, tiene presentación esporádica. La alteración básica en el síndrome de Marfan tipo 1 radica en una alteración en el gen de la fibrilina (FBN1), en el cromosoma 15 (15q21.1). La fibrilina es una proteína del tejido conectivo, un constituyente de elasticidad tisular y muy abundante en los tejidos afectados en los pacientes con síndrome de Marfan, incluyendo la aorta, el ligamento suspensorio del cristalino y el periostio. Esto último sugiere la posibilidad de que el tejido conectivo/elástico normal en el periostio es necesario para prevenir la elongación de los huesos.
Existe un marcado grado de variabilidad clínica, tanto intrafamiliar como interfamiliar, para lo que no existe una explicación clara. La deficiencia en fibrilina 1 está asociada con un exceso de señalización del factor de crecimiento transformador β (TGF-β). La inhibición de TGF-β atenúa las manifestaciones clínicas de la enfermedad. Los antagonistas de TGF-β (bloqueante del receptor de angiotensina II, losartan) han demostrado un gran éxito en la mejora y prevención de varias de las manifestaciones clínicas del síndrome de Marfan60,61. La pérdida de la fibrilina 1 por cualquier mecanismo y, el incremento de la biodisponibilidad del TGF-β, pueden ser relevantes en el desarrollo del síndrome de Marfan62.
En una minoría de casos de fenotipo de síndrome de Marfan no se descubren mutaciones en el gen FBN1. En 2004 se identificaron por primera vez mutaciones en el gen TGFBR2 (síndrome de Marfan tipo 2), que pueden ser responsables del 10% de los casos61. En algunas familias se ha observado un patrón de herencia autosómico dominante para diferentes mutaciones en el gen TGFBR2. La identificación de mutaciones en el gen TGFBR2 en el síndrome de Marfan tipo 2 proporcionó una evidencia directa de la relación entre FBN1 y TGF-β en humanos.
Se han descrito más de 500 mutaciones en FBN1 en el síndrome de Marfan63; por el contrario, el número de mutaciones en TGFBR2 es todavía muy limitado.
Para el diagnóstico clínico, aún deben seguirse los criterios revisados en 1996 (Ghent Nosology)64. Ellos se basan en los cuatro hallazgos más importantes:
1) esqueleto, 2) ojo, 3) sistema cardiovascular, y 4) ectasia dural del canal espinal lumbosacro. El diagnóstico del síndrome de Marfan puede efectuarse en un caso índice cuando: se evidencia una afectación severa en dos sistemas orgánicos diferentes y una afectación menor de un tercero, o cuando existe una mutación en el gen FBN1, una afectación severa de un sistema orgánico y una menor de un segundo sistema.
Para un pariente de un caso índice, el diagnóstico puede hacerse cuando existe un criterio mayor proporcionado por la historia familiar y otro criterio mayor por la afectación de un sistema orgánico, y afectación de un segundo sistema orgánico.
En un paciente sin historia familiar, en el que se hayan detectado mutaciones de la fibrilina 1, se necesitan, además, dos manifestaciones mayores en dos sistemas orgánicos para poder efectuar el diagnóstico de síndrome de Marfan. Téngase en consideración que un paciente puede padecer una «fibrilinopatía» sin ser un síndrome de Marfan.
Mujeres y hombres con síndrome de Marfan pueden presentar talla muy alta. El tratamiento para reducirla puede beneficiarles. Además, dado que la escoliosis y la cifosis pueden desarrollarse hasta en un 60% de estos pacientes, detener su crecimiento puede ser muy importante. No obstante, debe prestarse una gran atención al posible desarrollo, prevención y tratamiento de las anomalías cardiovasculares. Así, el propranolol puede reducir la fracción de eyección ventricular de la aorta ascendente y, en consecuencia, se emplea de forma rutinaria en cualquiera de estos pacientes que haya iniciado dilatación de la raíz aórtica. Con la experiencia adquirida en los modelos de ratón, debe añadirse un bloqueante del receptor de angiotensina II. Finalmente, reemplazar la aorta ascendente y la válvula aórtica e intervenir el aneurisma de la raíz aórtica ha incrementado sustancialmente la esperanza de vida de estos pacientes que son adecuadamente diagnosticados y convenientemente tratados60.
Síndrome de Beals [contractura racnodactílica congénita –CCA-]Las alteraciones esqueléticas son similares a los del síndrome de Marfan, con dolicostenomelia y aracnodactilia, con campidactilia en los dedos de la mano. La diferencia con el síndrome de Marfan radica en que existen contracturas articulares. El hélix del lóbulo de la oreja se encuentra plegado (fig. 5). La cifosis, escoliosis o cifoescoliosis se encuentran presentes en el 50% de los casos.

Otras manifestaciones clínicas incluyen: micrognatia, anomalías septales auriculares y ventriculares. En varios pacientes se ha diagnosticado prolapso mitral65, anomalías cardíacas estructurales y, ocasionalmente, dilatación de la raíz aórtica similar a la descrita en el síndrome de Marfan y ectopia del cristalino66. Todo ello sugiere que estos pacientes necesitan controles oftalmológicos y ecocardiográficos periódicos.
La causa del síndrome de Beals radica en mutaciones de un segundo gen de la fibrilina (FBN2) cartografiado en 5q23-31. Se hereda siguiendo un patrón autosómico dominante.
El diagnóstico se basa en los hallazgos clínicos. Si bien se trata de una entidad rara, se han descrito al menos 33 árboles genealógicos con CCA. Estos pacientes pueden presentar talla alta, pero aún no se dispone de experiencia en el tratamiento de la misma.
FibrilinopatíasExiste un amplio grado de variabilidad clínica en y entre las familias con síndrome de Marfan, así como entre los sujetos con enfermedades relacionadas con el tejido conectivo.
El término «fibrilinopatías» se aplica a las entidades clínicas asociadas con anomalías de la fibrilina 1 o fibrilina 2. En ellas se incluye el previamente descrito síndrome de Marfan tipo 1 y la contractura aracnodactílica congénita (FBN2), así como otras enfermedades severas del tejido conectivo causadas por mutaciones en el gen FBN1, que no reúnen los criterios para poder considerarse un síndrome de Marfan67 como: la ectopia del cristalino dominante, los aneurismas aórticos ascendentes68, las anomalías esqueléticas aisladas de síndrome de Marfan sin manifestaciones oculares ni cardiovasculares69 y la craneosinostosis marfanoide o síndrome de Shprintzen-Goldberg70.
Las mutaciones del gen de fibrilina se encuentran a lo largo de todo el gen y, con la excepción del síndrome de Marfan neonatal, no se demuestra asociación fenotípica.
Las mutaciones en el gen de la fibrilina 1, que alteran el procesamiento de la profibrilina, pueden dar lugar a anomalías esqueléticas del síndrome de Marfan en algunas familias. En una publicación, seis individuos de la familia del probando presentaban la mutación en el gen de la fibrilina 1, cosegregando con el fenotipo de talla alta. Ninguno de los sujetos tenía anomalías cardíacas ni oculares. Por esta razón, los autores sugieren que el gen de la fibrilina 1 es uno de los que determinan la talla en la población general69.
Por consiguiente, debe tenerse en mente que algún paciente con talla alta pudiera tener una mutacion en el gen FBN1.
Homocistinuria tipo 1La homocistinuria es un error congénito del metabolismo debido a una deficiencia en la enzima cistationina β sintetasa (CBS), descrito inicialmente por Carson et al y Gerritsen y Waisman en 1963. La CBS convierte la homocisteína en cistationina, reacción catalizada por la vitamina B6, actuando como cofactor. Su prevalencia se estima en torno a 1:200.000 nacidos vivos; sin embargo, en Irlanda, es de 1:40.000 nacidos vivos. Aproximadamente el 40% de los pacientes responden a altas dosis de vitamina B6 (piridoxina), habitualmente con manifestaciones clínicas más moderadas que los que no responden al tratamiento con vitamina 671. La falta de respuesta a esta vitamina requiere la administración de ácido fólico, a la dosis de 1 a 5mg cada 24 horas.
La deficiencia en CBS se hereda de forma recesiva, existiendo una heterogeneidad genética considerable en estos pacientes. El gen de la CBS (CBS) ha sido cartografiado en 21q21.
Las manifestaciones clínicas son similares a las del síndrome de Marfan, con algunas diferencias72. En estos pacientes, suelen afectarse cuatro sistemas orgánicos: ocular, esquelético, vascular y sistema nervioso central.
La ectopia o subluxación hacia abajo del cristalino (contraria a la dislocación hacia arriba del mismo en los pacientes con síndrome de Marfan) es el hallazgo más consistente. Las anomalías esqueléticas muestran un fenotipo similar al de los pacientes con síndrome de Marfan, con dolicostenomelia (brazos y piernas largos y delgados), aracnodactilia y talla alta, a menudo con proporciones eunucoides. Entre las anomalías esqueléticas más consistentes, se encuentra la osteoporosis. La escoliosis y la cifosis acontecen con frecuencia. Otras alteraciones incluyen: genu valgum y pectus carinarum o excavatum (fig. 6). La alteración más frecuente del sistema nervioso central es el retraso mental (en torno al 50% de los pacientes presentan un nivel intelectual entre 30 y 75). Pueden presentar episodios tromboembólicos que afectan a vasos grandes y pequeños a lo largo de la vida, particularmente en el cerebro. Estos episodios ocurren en el 6% de los pacientes.

Los grupos sulfidrilo de la homocisteína interfieren con el colágeno, causando anomalías del mismo. Dadas las similitudes de muchos de los elementos clínicos de la homocistinuria y del síndrome de Marfan, es razonable creer que muchas de sus manifestaciones, incluyendo la talla alta, estén relacionadas con cambios cualitativos de fibrilina. Los grupos sulfidrilo pueden también contribuir a la alteración del endotelio vascular y, en consecuencia, a la trombosis.
Síndrome de Beckwith-Wiedemann (BWS)Inicialmente fue descrito de forma independiente por Beckwith y Wiedemann en 1963. Está asociado con hipercrecimiento prenatal y posnatal, presentando las características clínicas más llamativas al nacimiento: onfalocele o anomalías umbilicales, macroglosia, lóbulo de la oreja arrugado y gigantismo (figs. 7 y 8). Entre el 30 y el 50% presentan episodios de hipoglucemia severa persistente, que se inician en los primeros días de vida, debidos a la existencia de hiperinsulinismo, como resultado de hiperplasia de las células de los islotes pancreáticos. Asimismo, muestran visceromegalias, con aumento del hígado, riñones, páncreas y, en ocasiones, del corazón. De forma consistente, presentan displasia medular renal, citomegalia córticosuprarrenal fetal e hipoplasia de las células intersticiales de las gónadas. La hemihipertrofia está presente en el 12,5% de los casos73,74.
. La macroglosia es todavía evidente. No presentaba hipoglucemias.")

Existe un incremento en la incidencia de tumores malignos (7,4 a 10%), siendo el más frecuente el tumor de Wilms y el carcinoma córticosuprarrenal. Otros tumores descritos son: nefroblastoma, hepatoblastoma y rabdomiosarcoma75.
Los niños con síndrome de Beckwith-Wiedemann son grandes al nacimiento. La velocidad de crecimiento se sitúa por encima del percentil 90 hasta los 4-6 años de edad, siendo normal posteriormente. Suelen alcanzar una altura media de 2,5 DE en el momento de desarrollar la pubertad, con un peso que oscila entre el percentil 75 y 90.
La prevalencia estimada se sitúa en torno a 1:14.000 personas. Aproximadamente el 85% de los casos son esporádicos y, el 15% heredados, sugiriendo un patrón de herencia autosómico dominante con penetrancia incompleta.
Hoy se acepta que este síndrome es una alteración genética compleja y heterogénea debida a anomalías en la impronta de genes relacionados con el crecimiento y el ciclo celular en la región cromosómica 11p1576,77. Estos genes agrupados son regulados de forma diferencial por regiones metiladas o por dominios que afectan a la actividad genética. La señal de impronta primaria para cada uno de estos genes es la metilación de ADN78.
Se conocen dos centros de impronta (IC) o dominios en el cromosoma11p15. IC1 se sitúa próximo al telómero y contiene dos genes (IGF2 y H19; este último, expresado por la madre y con impronta paterna, que puede servir como un supresor tumoral). Normalmente, el H19 paterno está metilado y silenciado, permitiendo la expresión paterna del gen adyacente IGF2, encontrándose el H19 materno expresado y el IGF2 materno silenciado. Cuando el gen materno H19 está metilado, su expresión está inhibida, mientras que la de IGF2 no se encuentra reprimida y da lugar a su expresión bialélica79.
La pérdida de impronta del gen materno IGF2 es una de las alteraciones moleculares más frecuentemente detectadas en pacientes con síndrome de Beckwith-Wiedeman, sin anomalías cromosómicas. La sobreexpresión de IGF2 puede también surgir de la disomía uniparental paterna del cromosoma 11 o de duplicaciones de la región cromosómica paterna11p15 asociada con trisomía del11p.
El centro de impronta o dominio 2 (IC2) es centromérico en relación a IC1, conteniendo varios genes. CDKN1C es un gen expresado maternalmente y con impronta paterna. Aproximadamente de 5 a 10% de los pacientes esporádicos y un 40% de los casos familiares con BWS presentan mutaciones en CDKN1C, dando lugar a expresión bialélica de CDKN1C. KCNQ10T1 es un gen expresado paternalmente y con impronta materna. La pérdida de metilación del gen materno KCNQ10T1 con expresión bialélica del gen KCNQ10T1 se aprecia en el 50 al 60% de los casos esporádicos.
IGF-II es esencial para el crecimiento fetal. Los experimentos transgénicos en ratones han mostrado que la pérdida del alelo funcional paterno igf2 acontece en el 40% de los casos de retraso de crecimiento prenatal80; la sobreexpresión de igf2 puede ser responable de la mayoría de los síntomas del BWS81,82 y, la alteración del igf2r genera hipercrecimiento y niveles elevados de IGF-II83. El hipercrecimiento en el síndrome de Beckwith-Wiedemann se produce en órganos ricos en IGF-II. El receptor de IGF-I se activa por IGF-I e IGF-II, siendo ambos los reguladores más importantes de la proliferación celular y el crecimiento somático. La sobreexpresión de IGF2 conduce al hipercrecimiento pre y posnatal. Además, la impronta de IGF2 se pierde en tumores esporádicos de Wilms, sugiriendo que la sobreexpresión de IGF2 es responsable del hipercrecimiento y del desarrollo de los tumores84.
El diagnóstico se basa en las manifestaciones clínicas descritas. Algunos de los cambios fenotípicos son similares a los de los hijos de madres diabéticas y a los de los pacientes con síndrome de Simpson-Golabi-Behmel syndrome (SGBS).
La detección y el tratamiento de las hipoglucemias son de gran importancia para la supervivencia y para prevenir daño neurológico. Estos pacientes requieren seguimiento periódico para el posible desarrollo de procesos tumorales. El control ecográfico de los riñones debe efectuarse cada 3 meses durante los 6 primeros años, ya que el tumor de Wilms y el carcinoma córticosuprarrenal son las neoplasias más frecuentes en estos pacientes. Los pacientes con hemihipertrofia y disomía uniparental tienen una alta frecuencia de procesos tumorales (hepatoblastomas y tumor de Wilms).
Hipercrecimiento somático (metilción y silenciamiento de H19)El gen H19, cuyo locus se localiza en la región 11p15, está estrechamente relacionado con el gen IGF2 en el IC1. Estos dos están imprintados de forma opuesta. La región diferencialmente metilada (DMR) controla la expresión del alelo específico de los genes H19 e IGF2. H19 tiene impronta paterna y se expresa el de origen materno en la mayoría de los tejidos. La expresión bialélica de igf2 por alteración de la impronta materna de igf2 ha sido demostrada en el ratón, como resultado de un gen H19 heredado de la madre. Asimismo, también se ha descrito en el síndrome de Beckwith-Wiedemann y en los tumores de Wilms la expresión, bialélica de IGF2, asociada con metilación y silenciamiento del gen H19 expresado normalmente por la madre.
Morison et al analizaron la expresión de la metilación de H19 e IGF2 en niños con hipercrecimiento, sin diagnóstico clínico de BWS, sugiriendo un síndrome particular85. En tres de los seis niños con hipercrecimiento somático, existía metilación anormal y silenciamiento del H19 y sobreexpresión de IGF2. Esta observación tiene implicaciones relevantes para la evaluación de los niños con hipercrecimiento sin otras manifestaciones sugestivas de un síndrome.
Síndrome de Simpson-Golabi-Behmel (SGBS), tipos 1 y 2Fue descrito inicialmente por Simpson en 1975, y por Golabi y Behmel, independientemente, en 1984. El gen fue identificado por Pilia en 1996.
Se hereda siguiendo un patrón recesivo ligado al X. Se caracteriza por hipercrecimiento pre y posnatal, con apariencia facial inusual (descrito como síndrome «bulldog»), digital y otras anomalías. Las mujeres portadoras pueden tener algunos cambios faciales.
Los varones afectados pueden alcanzar tallas adultas entre 192 y 210cm. Presentan macrosomía, visceromegalia, onfalocele, displasisa renal, plegamiento del lóbulo de la oreja, hipoglucemia neonatal por hiperplasia de las células de los islotes pancreáticos y riesgo de tumores embrionarios (tumor de Wilms, neuroblastoma y carcinoma hepatocelular en la infancia temprana), elementos similares a los del BWS. La mortalidad perinatal e infantil es alta86,87.
Cursan con hipercrecimiento, cambios faciales características (mandíbula grande y protruyente, punta nasal hacia arriba, puente nasal amplio, nariz ancha, boca grande, lengua grande y labios gruesos), paladar arqueado alto o labio palatino, macrocefalia, uñas de los dedos índice ausentes o hipoplásicas y hernia inguinal. La edad ósea se encuentra acelerada. La inteligencia es normal y, en ocasiones, está ligeramente retrasada. La hipotonía es habitual.
Este síndrome es el resultado de diferentes mutaciones o microdeleciones del gen del glipicano-3 (GPC3) en Xq26 (SGBS1), sugiriendo que SGBS es debido a la existencia de una proteína GPC-3 no funcional. GPC-3 interactúa con IGF-II y puede ser importante en la modulación de IGF2. En la actualidad, se cree que el glipicano controla el crecimiento del tejido embrionario mesodérmico, actuando en conjunción con IGF2.
Los estudios en ratón con doble mutación indican que la proteína glipicano 3 no secuestra IGF-II o IGF-I, sugiriendo que los efectos de glipicano 3 e IGF sobre la proliferación celular y el crecimiento somático convergen en un lugar específico «aguas abajo».
Las mutaciones inactivantes de GPC3 conllevan crecimiento excesivo. De ello puede inferirse que la función normal del glipicano 3 es la de limitar la proliferación celular. El ratón deficiente de gpc3 presenta alteraciones típicas del SGBS. El hipercrecimiento del ratón gpc3 es similar al del ratón deficiente en el receptor de IGF-II (igf2r), un regulador negativo de IGF-II88. Teóricamente, la proteína glipicano 3 podría facilitar la degradación de IGF-II a través de su interacción con el receptor de IGF-II.
Los pacientes con SGBS tienen muchos aspectos de solapamiento con pacientes afectados de síndrome de Beckwith-Wiedemann y, en algunos casos, los estudios moleculares son imprescindibles para efectuar un diagnóstico correcto. Este solapamiento clínico puede ser debido a la sobreexpresión de IGF-II.
Brzustowicz et al89 cartografiaron un locus en Xp22, asociado con SGBS tipo 2. El gen GPC3 está excluido de esta región.
Síndrome PTEN hamartomaHeredado según un patrón autosómico dominante, incluye el síndrome de Bannayan-Riley-Ruvalcaba (BRRS), la enfermedad de Cowden y la enfermedad de Lhermitte-Duclos (LDD)90.
«Phosphatase tensin homolog in chromosome 10» (PTEN ) codifica una proteína con actividad supresora tumoral que defosforila tanto substratos lipídicos como proteicos y regula negativamente la vía de señalización estimulada por factores de crecimiento como IGF-I, controlando indirectamente el ciclo celular y la apoptosis91. PTEN desempeña una función relevante en procesos malignos en el ser humano. Las mutaciones y deleciones en PTEN se han observado en casos esporádicos de cáncer de mama, cerebro, próstata y riñón (en líneas celulares) y en diversos tumores primarios, como carcinoma endometrial, melanoma maligno y tumores de tiroides.
Las mutaciones de la línea germinal de PTEN se han identificado en el 60% de los pacientes con síndrome de Bannayan-Riley-Ruvalcaba (BRRS), en el 80% de pacientes con enfermedad de Cowden y en pacientes con enfermedad de Lhermitte-Duclos (LDD) (OMIM 158350). En la actualidad, se considera que estas tres entidades clínicas son alteraciones alélicas heterocigotas, constituyendo probablemente la misma enfermedad.
Los pacientes pueden tener elementos clínicos característicos de una u otra entidad, presentando mutaciones idénticas de PTEN. Se han descrito miembros de una misma familia con manifestaciones clínicas de BRRS y de enfermedad de Cowden o de enfermedad de Cowden y LDD. Además, algunos pacientes con BRRS pueden desarrollar características de enfermedad de Cowden con la edad92.
El síndrome BRRS cursa con macrocefalia, múltiples hamartomas (lipomas, hemangiomas, linfangiomas, pólipos intestinales) y otros tumores (seminoma, germinoma), macrosomía al nacimiento, manchas pigmentadas en cuerpo y el glande del pene (fig. 9) y pseudopapiledema46,73.

La enfermedad de Cowden se ha descrito fundamentalmente en adultos. El patrón de herencia es autosómico dominante y cursa con macrocefalia, múltiples lesiones hamartomatosas, especialmente de la piel y de las membranas mucosas, con lesiones verrucosas en la piel de la cara y las extremidades, así como pápulas similares a un empedrado de las encías y la mucosa bucal, pero también hamartomas y neoplasias de los órganos internos, siendo más frecuentes en tiroides, mama y ovario46,92.
La macrocefalia progresiva, la lengua escrotal y el retraso mental de discreto a moderado, son signos importantes en los lactantes. Los triquilemomas en los pliegues nasolabiales y los pliegues palmares y plantares hiperqueratósicos son habitualmente visibles en el niño mayor, acompañándose a menudo de lipomas subcutáneos y hemangiomas cutáneos.
La enfermedad de Lhermitte-Duclos (LDD) o gangliocitoma displásico del cerebelo, conocido previamente como enfermedad parenquimatosa del cerebelo VI (OMIM #601728), es una lesión cerebelosa rara, que puede acontecer esporádicamente o en asociación con la enfermedad de Cowden93,94. El comienzo en la época adulta de la enfermedad de Lhermitte-Duclos es ahora considerado patognomónico de enfermedad de Cowden. Se han descrito aproximadamente 220 casos de LDD.
La resonancia magnética en pacientes con LDD es habitualmente diagnóstica94. Se caracteriza por macrocefalia (que puede ser progresiva), retraso en el desarrollo, convulsiones, signos cerebelosos (temblores, disdiadocoquinesia) y, en algunos casos, hipertensión intracraneal, como consecuencia de la herniación de las amígdalas cerebelosas. Existe una hipertrofia global del cerebelo, giros rizados, patrón de córtex invertido e hipercrecimiento hamartomatoso por las células ganglionares hipertróficas, que reemplazan las células granulares y las células de Purkinje del cerebelo.
Las mutaciones de PTEN pueden también encontrarse hasta en el 20% de casos de síndrome de Proteus y, aproximadamente, en el 50% de los casos de síndrome Proteus-like90.
Dada la variedad de hamartomas y tumores en los pacientes con BRRS, enfermedad de Cowden y enfermedad de LDD, que pueden presentarse a cualquier edad92, se necesita un largo y amplio seguimiento de los mismos. El tratamiento depende del tipo de hamartoma o tumor.
Síndrome de PartingtonEn 1997 Partington et al95 publicaron 3 familias con 11 miembros, varones y mujeres, que mostraban un síndrome de hipercrecimiento, discreto o moderado retraso mental y duplicación del, 4p16.3. Los tejidos blandos se encuentran afectados, con engrosamiento de la cara. El cabello de la cabeza es abundante y las cejas son grandes con pelo abundante. El perímetro craneal es grande, arcos supraorbitarios prominentes y mandíbula cuadrada, con nariz pequeña. Las manos y los pies son grandes. Existe una variación considerable en las alteraciones presentes en este síndrome entre los distintos miembros de una misma familia.
La talla se sitúa por encima de la media. Aunque el hipercrecimiento puede estar presente en el lactante, es más evidente en el adolescente (todos se sitúan por encima del percentil 75) y en el adulto joven. La media de altura en el adulto varón se sitúa en torno a 187,75cm con un rango de 181cm a 197cm y para la mujer adulta de 171,1cm con un rango de 166cm to 179cm.
La región 4p16 contiene diversos genes, algunos de los cuales son responsables de: enfermedad de Huntington, acondroplasia, displasia tanatofórica e hipocondroplasia.
Las tres familias publicadas por Partington et al presentan translocaciones diferentes. Los cuatro sujetos con hipercrecimiento recibieron una translocación no balanceada con tres copias de 4p16.3 y un miembro recibió solo una copia de 4p16.3 y presentó talla baja. Los autores sugieren que FGFR3 podría ser un candidato para las anomalías del crecimiemnto (3 dosis conllevarían hipercrecimiento y 1 única dosis, hipocrecimiento–síndrome de Pitt-Rogers-Danks -).
La sugerencia de que dosis adicionales de FGFR3 pudieran ser responsables de hipercrecimiento, está en debate, dado que existen evidencias que indican que FGFR3 es un regulador negativo del crecimiento óseo. Por consiguiente, se requieren estudios adicionales para saber si el síndrome descrito por Partington et al es el resultado de una sobredosis del gen FGFR3, mutaciones inactivantes del gen u otros genes en 4p16.3.
Síndrome de SotosDescrito por primera vez en 196496 (fig. 10). Los factores diagnósticos más importantes incluyen: configuración facial característica, gran dolicocefalia craneal, hipercrecimiento y enfermedad neurológica no progresiva con retraso mental. La prevalencia es desconocida, pero probablemente es uno de los síndromes de hipercrecimiento más frecuentes después del BWS y el síndrome de Marfan. Los varones y las mujeres se afectan de forma similar.
, con braza de 173cm, manos y pies grandes, dolicocefalia (59cm de perímetro craneal), frente agrandada, fisuras palpebrales hacia abajo, orejas grandes, mandíbula prominente, escoliosis dorsal moderada, con coeficiente intelectual de 70.")
Paciente original que describiera el síndrome de Sotos: varón de 10 años y 3 meses, con talla de 156cm (similar a un varón de 14 años y medio), con braza de 173cm, manos y pies grandes, dolicocefalia (59cm de perímetro craneal), frente agrandada, fisuras palpebrales hacia abajo, orejas grandes, mandíbula prominente, escoliosis dorsal moderada, con coeficiente intelectual de 70.
El hipercrecimiento es prenatal y posnatal. La velocidad de crecimiento es particularmente excesiva entre los 3 y 4 años de edad, decelerándose posteriormente, pero en los percentiles altos. Las tallas adultas en varones se sitúan en torno a 186±5,7cm y, en mujeres en torno a 173,1±7,7cm)97, si bien algunos individuos pueden alcanzar tallas superiores.
La configuración craneofacial es muy característica: frente prominente, entradas en el cabello frontoparietal, dolicocefalia, hipertelorismo, fisuras palpebrales inclinadas hacia abajo, paladar arqueado alto y estrecho, con irregularidades y prominencia palatina y mentón punteaguado (figs. 10–12). Erupción prematura de los dientes en el 60 al 80% de los casos y edad ósea avanzada en el 75 al 84% de los pacientes.
, frente agrandada, fisuras palpebrales hacia abajo, paladar arquedado alto, mentón punteagudo y orejas grandes. Mostraba varios meses de retraso en el desarrollo mental.")
Niña de 2 años con talla en P95 y características craneofaciales típicas de síndrome de Sotos: dolicocefalia (perímetro cefálico de 55cm, equivalente a una niña de 6 años), frente agrandada, fisuras palpebrales hacia abajo, paladar arquedado alto, mentón punteagudo y orejas grandes. Mostraba varios meses de retraso en el desarrollo mental.
, el cual también se encuentra afectado de síndrome de Sotos, igual que su hermanastra (no aparece en la fotografía). Nótese en ambos las características craneofaciales de este síndrome.")
Niña de 4 años y 8 meses con síndrome de Sotos. La talla adulta fue de 188cm. A la derecha, padre de la paciente (talla de 198cm), el cual también se encuentra afectado de síndrome de Sotos, igual que su hermanastra (no aparece en la fotografía). Nótese en ambos las características craneofaciales de este síndrome.
Las manifestaciones clínicas del sistema nervioso central son frecuentes, incluyendo: retraso en el desarrollo, retraso en el inicio y forma de caminar y hablar y torpeza (60 a 80% de los casos), así como hipotonía y articulaciones laxas. La deficiencia mental está presente en el 85 al 100% de los pacientes, con una media de coeficiente intelectual de 75 y un rango de 40 a moderadamente retrasado. Del 10 al 15% pueden ser mentalmente normales. El 30% pueden presentar convulsiones. Existe un incremento de incidencia de desarrollo tumoral (∼2,2 a 3,9%). Se han descrito anomalías de los ventrículos e incremento de los espacios subaracnoideos, así como alteraciones cardíacas, musculoesqueléticas (cifoescoliosis), urogenitales (hidronefrosis, riñones hipoplásicos, agenesia renal, dilatación de la pelvis renal), hernias inguinales, alteraciones oftalmológicas (estrabismo, alteraciones refractarias, alteraciones del nervio óptico o de la retina), endocrinopatías (hipotiroidismo primario) y otras. Hasta la fecha, no se ha demostrado ninguna alteración endocrinológica que explique su hipercrecimiento46.
Desde hace años se sospecha que la causa de este síndrome sea de origen genético, habiéndose descrito varias familias con miembros afectos, que sugieren un patrón de herencia autosómico dominante98. No obstante, aunque la mayoría de los casos son esporádicos (85%), podrían deberse a mutaciones nuevas.
En 2002 Kurotaki et al99 identificaron deleciones o mutaciones puntuales en un único gen: «nuclear receptor binding SET domain protein 1» (NSD1) en 5q35, en el 75% de los casos esporádicos, indicando que la haploinsuficiencia de NSD1 es la causa más importante del síndrome de Sotos. El hallazgo de que todas las mutaciones encontradas en NSD1 eran heterocigotas o hemicigotas es consistente con un patrón autosómico dominante. Hoy sabemos que existen diferencias entre las anomalías en NSD1 entre pacientes japoneses y pacientes no japoneses. La incidencia de las deleciones es más elevada en pacientes japoneses, mientras que la presencia de mutaciones intragénicas es más común en pacientes no japoneses. Los pacientes con microdeleciones tienen más dificultades en el aprendizaje que los pacientes con mutaciones100.
El gen NSD1 se expresa en el cerebro fetal, lo que podría explicar el gran tamaño cerebral y la deficiencia mental. Además, también se expresa en músculo esquelético, riñón, bazo, leucocitos y timo. La proteína NSD1 interactúa con el dominio de unión de los receptores hormonales nucleares, pudiendo actuar como corepresor o como coactivador. El hallazgo de que la haploinsuficiencia de NSD1 induce hipercrecimiento en el síndrome de Sotos, implica que el gen NSD1 actúa como corepresor de genes del crecimiento. La descripción de un paciente con una duplicación de 5q35.2,q35.3, que presentaba microcefalia y talla baja, sugiere un efecto dosis del gen NSD1 causando un fenotipo inverso al del síndrome de Sotos101.
NSD1 es un miembro de una familia de proteínas que incluye NSD2 y NSD3, con dominios de funciones similares y 70 a 75% de secuencia idéntica con NSD1. No se ha demostrado ninguna mutación ni deleciones en NDS2 ni en NSD3 en pacientes con síndromes de hipercrecimiento102.
Un análisis de correlación genotipo-fenotipo fue efectuado en 266 pacientes con anomalías de NSD1100. El 93% de ellos, que habían sido diagnosticados clínicamente de síndrome de Sotos, tenían mutaciones en NSD1, de las que el 83% eran mutaciones intragénicas y, el 10%, microdeleciones. Es posible que el 7% de pacientes en los que no se detectaron mutaciones las tuvieran, pero no se identificaran. La configuración de las características faciales estuvo presente en el 99% de los casos, la alteración en el aprendizaje en el 97% y, el hipercrecimiento, en el 90% de ellos. La edad ósea avanzada estuvo presente en el 76% de los pacientes.
Otras correlaciones genotipo-fenotipo, con variaciones menores, han sido detectadas por otros autores103-106.
Los estudios moleculares han confirmado las observaciones efectuadas hace 47 años96, de un síndrome con las características fundamentales de dolicocefalia, configuración facial específica, hipercrecimiento y alteraciones neurológicas no progresivas con retraso mental.
No existe ningún marcador bioquímico para el diagnóstico de este síndrome. El diagnóstico de sospecha debe basarse en los datos clínicos. El diagnóstico de confirmación requiere estudios moleculares. Algunos test diagnósticos, como el del cromosoma X frágil, la realización de un cariotipo o la determinación de los niveles séricos de GH e IGF-I son, preferentemente, para excluir otras entidades nosológicas. La resonancia magnética craneal ayuda a evidenciar las alteraciones ventriculares y de los espacios subaracnoideos, pero no es diagnóstica del síndrome de Sotos.
En relación al tratamiento, las niñas con una predicción de talla adulta superior a 178cm pueden beneficiarse de altas dosis de estrógenos para reducir su talla adulta. Las dificultades sociales y de conducta pueden requerir consejo psicológico. En cuanto al consejo genético, los individuos afectos tendrán un 50% de posibilidades de tener hijos afectos. Las dificultades de comportamiento y conducta durante la infancia, así como la inmadurez en la época adulta, pueden beneficiarse de consejo psicológico. Se debe estar permanentemente alerta ante el desarrollo de procesos tumorales (2,2- 3,9%) y al riesgo de transmisión. Dado que se hereda según un patrón autosómico dominante, los individuos afectos tienen un riesgo de 50% de tener hijos afectos.
En el caso de pacientes esporádicos, los padres sanos tendrán un riesgo de recurrencia <1%. Los sujetos afectos son fértiles. No existen evidencias, hasta el momento, de que su vida media esté reducida.
Síndrome de WeaverDescrito en 1974107, se caracteriza por hipercrecimiento prenatal o posnatal, facies inusual, maduración esquelética avanzada y camptodactilia.
Algunos pacientes varones han alcanzado una talla adulta de194,2cm, mientras que las mujeres adultas han alcanzado tallas de 176,3cm.
El hipercrecimiento se presenta al nacimiento o se inicia durante la lactancia. El 80% de los pacientes presentan desde retraso en el desarrollo hasta retraso mental (de medio a severo). Muchas de sus características recuerdan al síndrome de Sotos108.
Así, los pacientes con síndrome de Weaver presentan: hipertelorismo, orejas grandes, puente nasal deprimido e inclinación hacia abajo de las fisuras palpebrales; sin embargo, a menudo la dolicocefalia no está presente, siendo el occipucio plano. Además, no presentan mentón prominente y la cara es ancha. En muchos casos hay hipertonía más que hipotonía. La maduración ósea está avanzada, encontrándose más acelerada la edad del carpo que la de la mano. Los huesos largos son anchos y la camptodactilia es frecuente (fig. 13).

La mayoría de los casos son esporádicos, aunque el patrón de transmisión puede ser autosómico dominante. En 2003 se describieron las primeras mutaciones en el gen NSD1 en tres pacientes diagnosticados de síndrome de Weaver, sugiriendo que los síndromes de Sotos y Weaver eran, probablemente, alélicos109. Análisis posteriores en 2005 demostraron que estos tres pacientes tenían síndrome de Sotos y que ninguno de los pacientes con clásico síndrome de Weaver mostraba mutaciones en NSD1. Por todo ello, los autores sugirieron que el diagnóstico de síndrome de Weaver debe emitirse únicamente cuando se hayan excluido anomalías en el gen100. Su causa, por tanto, es aún desconocida.
Neurofibromatosis tipo I (NF1)–OMIM 162200La neurofibromatosis fue descrita inicialmente por von Recklinghausen. La neurofibromatosis tipo I (NF1) es una enfermedad frecuente que predispone al padecimiento de neoplasias en tejidos derivados de la cresta neural embrionaria, habiéndose descrito diferentes formas. La clásica o tipo von Recklinghausen ha sido denominada tipo I o NF1 (fig. 14).
 Neurofibroma de hemilengua izquierda. B) Fibromas en el dorso de un varón de 14 años de edad con neurofibromatosis tipo 1.")
Aunque la NF1 a menudo solo es una preocupación cosmética, en ocasiones puede presentar complicaciones graves y letales. No es posible predecir los síntomas que desarrollará cada paciente afecto de forma individualizada, si bien puede afirmarse que la prevalencia edad específica de la mayoría de los síntomas en pacientes con NF1 se incrementa con la edad110.
La neurofibromatosis tipo 1 (NF1) es debida a mutaciones en el gen NF1 en17q11.2, que codifica neurofibrina, una proteína relacionada con la regulación de la diferenciación y la proliferación celular. La neurofibrina se ha asociado con microtúbulos en algunos tipos celulares, actuando en otros tipos celulares como regulador negativo del protooncogen RAS, quién desempeña funciones moleculares críticas en muchas vías de señalización intracelular111.
La neurofibromatosis tipo I comprende, aproximadamente, el 90% de todos los casos de neurofibromatosis, siendo una de las anomalías autosómico dominantes más frecuentes, con una incidencia de 1 caso por cada 3.000 nacidos. Cerca del 50% de los casos son esporádicos.
La talla baja y la microcefalia no son infrecuentes. La macrocefalia puede estar presente en el 25 al 50% de los casos. Algunos pacientes (2 a 4%) presentan hipercrecimiento112. Algunos pueden presentar pubertad precoz central con incremento de su crecimiento. En algunos casos el gigantismo puede ser debido a una secreción excesiva de GH por la existencia de un glioma del nervio óptico o un tumor hipotalámico. La excesiva producción de GH puede deberse a una pérdida de inhibición somatostatinérgica de la secreción de GH por la invasión tumoral de las neuronas hipotalámicas112. Finalmente, en algunos pacientes, el hipercrecimiento acontece sin que se evidencie secreción excesiva de hormona de crecimiento.
Los pacientes con microdeleciones son típicamente más altos que aquellos con mutaciones intragénicas de NF1, sugiriendo que la deleción de un gen vecino tal vez promueva el crecimiento. Las mutaciones en el gen RNF135, el cual se sitúa en la región de la microdeleción de NF1, en 6 familias, demostró que la haploinsuficiencia de RNF135 contribuye al fenotipo de casos de microdeleciones de NF1113 con hipercrecimiento.
Un consenso del National Institute of Health (NIH)114 identificó 7 componentes importantes para el diagnóstico de NF1; a saber: 1. Presencia de 6 o más manchas café con leche de 5mm de diámetro o más en sujetos prepuberales y con un diámetro de más de 15mm en sujetos pospuberales; 2. Dos o más neurofibromas de nervios periféricos de cualquier tipo o un neurofibroma plexiforme; 3. Efélides en la región inguinal o axilar; 4. Glioma óptico; 5.Dos o más nódulos de Lisch (hamartomas del iris); 6. Presencia de una lesión ósea característica, como la displasia del esfenoides, en la base del cráneo, o pseudoartrosis o adelgazamiento del córtex, con o sin interrupción completa del hueso; 7. Un pariente de primer grado con NF1, de acuerdo con los criterios previos. Dos o más de estos componentes presentes permiten efectuar el diagnóstico de NF1.
El tratamiento del hipercrecimiento, si se necesita, es similar al referido para los niños y niñas con talla alta.
Síndrome de NevoAnomalía rara autosómica recesiva caracterizada por longitud incrementada al nacimiento, cifosis, hipotonía muscular y laxitud articular. Apenas se han publicado unos pocos casos. Algunos de ellos presentan características clínicas similares a los pacientes del tipo cifoescoliótico del síndrome de Ehlers-Danlos, tipo VI-A (EDS VIA), una anomalía heredada del tejido conectivo caracterizada por deficiencia de lisil-hidroxilasa debida a mutaciones en el gen PLOD1. Seis de siete pacientes con síndrome de Nevo mostraron una mutación puntual en el exón 9 del gen PLOD1 en homocigosis, que daba lugar a una mutación sin sentido, mientras que el séptimo paciente mostró una gran deleción en homocigosis en el exón 17 de PLOD1. Estos hallazgos sugieren que el síndrome de Nevo es alélico e indistinguible clínicamente del EDS VIA115.
Síndrome de Elejalde (OMIM 200995)También conocido como «displasia acrocefalopolidactílica», fue descrito por primera vez por Elejalde et al en 1977116. El elemento más importante es el gigantismo con pesos al nacimiento en dos pacientes publicados de 4,3 y 7,5Kg. Muestran un cuerpo redondeado, globular, con onfalocele, extremidades cortas, craneosinostosis, piel redundante en todo el cuerpo, pero fundamentalmente en el cuello. La craneosinostosis produce turricefalia. Muestran, asimismo, hipertelorismo, epicantus e inclinación palpebral hacia abajo117. El gigantismo puede detectarse en el precozmente mediante estudio ecográfico.
Uno de los hallazgos más importantes en estos pacientes es la excesiva cantidad de tejido conectivo en los riñones, páncreas, pulmones y piel, generando lesiones en los glomérulos, obstrucción y dilatación de los túbulos renales, enfermedad renal quística, hipoplasia pulmonar severa e hipoplasia e insuficiencia pancreática.
La otra característica de interés es el hipercrecimiento de las fibras nerviosas perivasculares en muchos de los tejidos.
Se han descrito algunos casos en familias consanguíneas, sugiriendo un patrón de herencia autosómico recesivo. Se ha hipotetizado que este síndrome podría estar asociado con una mutación inactivante del gen FGFR118.
El pronóstico dependerá de la gravedad de la displasia renal y la hipoplasia pulmonar, pues cuando son muy graves, son incompatibles con la vida. No obstante, es posible que existan formas moderadas que no se diagnostican. Deberá pensarse en el diagnóstico de este síndrome en el caso de fetos afectos de enfermedad poliquística renal bilateral. Es posible que no se diagnostiquen los casos más moderados durante el periodo neonatal o de lactancia117.
Hipercrecimiento prenatalExisten diferentes anomalías asociadas con hipercrecimiento prenatal. Todas ellos han sido incluidas en la tabla 2. Algunas ya han sido descritas. Sus características al nacimiento se indican en la tabla 4.
Características al nacer de anomalías con hipercrecimiento prenatal (macrosomía)
| ENFERMEDAD | ANOMALÍAS | TAMAÑO CRANEAL | HIPOGLUCEMIA | HERENCIA | OTROS |
| Hijo de madre diabética | ↑ grasa y masa muscularDistrés respiratorioHiperbilirrubinemiaTrombosis de la vena renalPolicitemia | Normal | Precoz - 75%Transitoria | --- | CardiomegaliaEstenosis subaórticaSíndrome de colon izquierdoAgenesia lumbosacra |
| Lactantes gigantes | Ninguna | Pequeño | Severa-persistente(hiperinsulinismo) | Esporádica (mayoría)Otras: AR o AD | --- |
| S. Beckwith-Wiedemann | MacroglosiaOnfaloceleOjos prominentesVisceromegaliaPliegue del lóbulo de la oreja | Pequeño | Severa-persistente(hiperinsulinismo) | Esporádica: 85%Familiar: 15% | Displasia renalHemihipertrofiaCitomegalia del córtex adrenalTumores: Wilms |
| S. Simpson-Golabi-Behmel | MacroglosiaVisceromegaliaHipertelorismoPliegue del lóbulo de la orejaFacies inusualHipotonía | Macrocefalia | Severa-persistente(hiperinsulinismo) | Ligada al XRecesiva | CardiopatíaUñas hipoplásicas de los dedos índice |
| S. Perlman | Abdomen distendidoEntradas en el cabelloLabio superior en V InvertidaMicrognatia | Normal | Severa(hiperplasia de células ß) | AutosómicaRecesiva | Displasia renalTumor de WilmsVisceromegalia:riñón-hígado |
| S. Sotos | Frente prominenteHipertelorismoMentón puntiagudoHipotonía | MacrocefaliaDolicocefalia | No | AutosómicaDominanteoEsporádica | Problemas de alimentaciónDificultad respiratoriaEdad ósea avanzada |
| S. Weaver | Frente prominenteHipertelorismo | Macrocefalia(occipucio plano) | No | Esporádica oAutosómica Dominante | Edad ósea muy avanzada |
| S. Nevo | Dorsiflexión de piésEdema | MacrocefaliaDolicocefalia | No | AutosómicaRecesiva | Edad ósea avanzadaPaladar alto arqueado |
| S. Marfan, neonatal, severo | Brazos y piernas largos y delgadosPérdida de piel redundante | NormalDolicocefalia | No | Esporádica oAutosómica Dominante | Enfisema pulmonar |
| Lipodistrofias | Grasa subcutánea disminuidaManos, piés y orejas grandes | Normal | No | AutosómicaRecesiva | Edad ósea avanzada |
| S. Elejalde | Fetos y recién nacidos dismórficosCuerpo globular redondeadoPolidactilia | Craneosinostosis(turricefalia) | No | ¿AutosómicaRecesiva? | Tejido conectivo excesivoRiñones poliquísticosHipoplasia pulmonar |
| S. Marshall-Smith | Ojos y frente prominenteEdad ósea muy avanzadaMetacarpianos y falanges proximales. y distales, anchas | Macrocefalia(dolicocefalia) | No | Esporádica | Dificultad respiratoria severa |
Los hijos de madre diabética son típicamente grandes, con pesos al nacimiento por encima de 2 DE para su edad gestacional y, con frecuencia, por encima de 4Kg. La longitud del paciente está más relacionada con el grado de hiperglucemia materno y el reflejo de la hiperglucemia e hiperinsulinemia fetal.
Los niños gigantes procedentes de madres normales, sin historia clínica de diabetes mellitus o intolerancia a la glucosa materna, tienen pesos al nacimiento que varían desde aproximadamente 3,8 a 5Kg.
Los pacientes afectos muestran hipoglucemia neonatal grave, como consecuencia del hiperinsulinismo, considerado causado en el pasado por la existencia de nesidioblastosis o síndrome de dismadurez de las células β. No suelen presentar malformaciones.
La mayoría de los casos son esporádicos. La posibilidad de que algunos de ellos sean variantes genéticas del síndrome de Beckwith-Wiedemann no puede excluirse, pues se han observado casos clínicos de síndrome de Beckwith-Wiedemann con hipercrecimiento sin malformaciones.
Algunos pueden presentar un adenoma único o múltiple de las células β de los islotes del páncreas, una causa infrecuente de hipoglucemia hiperinsulinémica en neonatos y lactantes.
Otra posibilidad es que estos niños gigantes estén afectos de una de las cinco formas anteriormente descritas de hiperinsulinismo cogénito4.
El síndrome de Perlman debe ser considerado en el diagnóstico diferencial del hipercrecimiento prenatal. Se caracteriza por gigantismo y hamatomas renales.
Consideraciones generalesLos cuadros clínicos de hipercrecimiento en el ser humano conforman un auténtico Tratado de Pediatría, en el que, desde las variantes de la normalidad, aún no bien entendidas, hasta las cromosomopatías, las entidades sindrómicas, la patología endocrinológica, la patología cardíaca, la patología oftalmológica, las anomalías del esqueleto, algunos errores congénitos del metabolismo, se han incrementado nuestros conocimientos de forma sustancial gracias a la aproximación genética, citogenética, epigenética y molecular.
Si bien la talla alta ha sido bien considerada socialmente, no es menos cierto que la reducción de la talla final se demanda con mayor frecuencia, preferentemente en mujeres y por razones de orden psicológico. Esta conducta de reducir la talla final es aún más aconsejable en los pacientes con síndrome de Marfan, síndrome de Sotos, síndrome de Klinefelter y otros cuadros en los que la predicción de talla y la repercusión sobre la estática del aparato locomotor lo haga aconsejable.
En los hipercrecimientos de origen endocrino el tratamiento es el correspondiente a la enfermedad que los origina.
Aunque el tratamiento con esteroides sexuales en general es bien tolerado, aún se requiere de más experiencia y valoración individualizada. Dicho tratamiento deberá evitarse en pacientes con la homocistinuria119 y otras situaciones con riesgo de trombosis.
Es obligado control clínico estrecho de todos los pacientes con cuadros de hipercrecimiento que conllevan un mayor riesgo de padecer ciertos tipos de tumores.
La realización de diagnóstico genético, con ulterior consejo genético, es obligada ante aquellas enfermedades o síndromes en los que hoy se conoce existe una base monogénica.
El conocimiento de nuevas enfermedades monogénicas que asocian talla alta y obesidad ha ampliado sustancialmente nuestros conocimientos, requiriendo una conducta médica diferente a la mantenida hasta hace pocos años19,120.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores desean expresar su particular agradecimiento a las personas que han colaborado desinteresadamente en permitir la reproducción de alguno de sus pacientes: Prof. Dr. Manuel Hernández Rodríguez, Dr. Ricardo Gracia Bouthelier y Dr. RHA Ruvalcaba. Los autores han solicitado el correspondiente permiso de publicación cuando el paciente estaba publicado.
