

Sitges, 17, 18 y 19 de mayo 2007
COMUNICACIONES ORALESCOMPARACIÓN DE LAS CONCENTRACIONES SÉRICAS DE ASPARAGINASA TRAS LA ADMINISTRACIÓN DE ASPARAGINASA E. COLIY DE ASPARAGINASA PEGILADA: ESTUDIO EN LOS PROTOCOLOS LAL SHOP-99 Y RECIDIVAS SHOP 2003I. Badell1, E. López1, R. Farré2, E. Zapico3, N. Pardo1, J. Estella4, J. Molina5, A. Navajas6, B. López-Ibor7 y J. Cubells1
1Servicios de Pediatría, 2Farmacia y 3Bioquímica. H. Sant Pau. Barcelona. 4H. Sant Joan de Déu. Esplugues. Barcelona. 5H. Virgen del Camino. Pamplona. 6H. de Cruces. Bilbao. 7H. Montepríncipe. Madrid. España.
Objetivo: La asparaginasa constituye uno de los pilares terapéuticos más importantes del tratamiento de la leucemia aguda linfoblástica infantil. En este trabajo se propone investigar la dosis necesaria de asparaginasa pegilada en las diferentes fases del protocolo, para alcanzar grados de actividad de asparaginasa útiles y similares a los obtenidos con la asparaginasa E. coli.
Material y métodos: Se incluyen 25 curvas de concentraciones plasmáticas efectuadas en 21 pacientes afectados de leucemia aguda linfoblástica, pertenecientes a cinco centros del país. Corresponden 13 curvas de asparaginasa E. coli a 8 pacientes que recibieron en la inducción 10 dosis de 15.000 U/m2 y a 5 pacientes que recibieron una dosis de 20.000-25.000 U/m2 en la fase de mantenimiento o en los bloques de intensificación. Las 12 curvas de asparaginasa pegilada corresponden a 6 dosis de 2.500 U/m2 y 6 dosis de 1.000 U/m2, administradas en diferentes fases del tratamiento. El motivo de la administración de asparaginasa pegilada fue la presentación de reacción a asparaginasa E. coli. La administración se realizó en todos los pacientes por vía intramuscular. Previo consentimiento informado, se procedió a la determinación de la actividad de asparaginasa en los días 0, 3, 6, 9, 12 y 15. La actividad de la asparaginasa se determinó por un método colorimétrico (Medac Asparaginase activity test, MAT), con un límite de detección de 30 U/l. La actividad de asparaginasa mínima y necesaria para conseguir una depleción de asparagina del 100% es de 100 U/l.
Resultados: Respecto a la asparaginasa E. coli, la actividad de asparaginasa fue adecuada hasta 12 días en los 13 pacientes que recibieron 10 dosis de asparaginasa E. coli e incluso hasta 15 días en 2 de ellos. La actividad de asparaginasa fue de 3 a 8 veces superior al mínimo necesario durante los 12 días. En los 5 pacientes que recibieron una sola dosis de asparaginasa, la actividad se mantuvo hasta 6 días y en uno de ellos hasta 9 días. Una paciente que presentó reacción alérgica, mostró precozmente disminución de la actividad, sugiriendo producción de anticuerpos. Con la asparaginasa pegilada, la actividad de asparaginasa se mantuvo hasta 12 días e incluso 15–18 días con una sola dosis de 2.500 U/m2. Con la dosis de 1.000-1.250 U/m2, se mantuvo la actividad hasta 6 días, e incluso 9 días en un paciente. Algunos pacientes mostraron desde el inicio actividad nula, lo que sugiere producción de anticuerpos.
Conclusiones: Una sola dosis intramuscular de asparaginasa pegilada de 2.500 U/m2 puede sustituir las 10 dosis intramusculares de asparaginasa E. coli de 15.000 U/m2 para mantener actividad de asparaginasa superior a 100 U/l durante un mínimo de 12 días. Asimismo, una sola dosis intramuscular de asparaginasa pegilada de 1.000-1.250 U/m2 puede sustituir una dosis intramuscular de asparaginasa E. coli de 20.000-25.000 U/m2 para mantener actividad de asparaginasa superior a 100 U/l durante un mínimo de 6 días.
CALIDAD DE VIDA EN JÓVENES SUPERVIVIENTES DE LEUCEMIA SOMETIDOS A TRASPLANTE DE MÉDULA ÓSEA Y SUPERVIVIENTES DE TUMORES ÓSEOSA. Robert Segarra1, M. Daniel Llach2, M. Torrent Español2, I. Clemente3, I. Badell Serra2, N. Pardo García2 y J. Nadal Amat2
1Servicio de Psiquiatría y 2Servicio de Pediatría. Hospital de la Santa Creu i Sant Pau. UAB. Barcelona. España. 3Department of Pediatrics. UCLA School of Medicine. Los Ángeles, California. EE.UU.
Objetivos: Los objetivos del presente estudio son, por un lado, valorar y comparar la calidad de vida (CV) en una muestra de supervivientes de leucemia infanto-juvenil sometidos a trasplante de médula ósea (TMO) frente a supervivientes de tumor óseo juvenil, y, por otro lado, analizar las variables sociodemográficas y/o clínicas relacionadas con la CV de que informan los supervivientes.
Material y método: La muestra se compone de 42 jóvenes mayores de 16 años que han sobrevivido a un cáncer diagnosticado en la infancia o adolescencia (leucemia o tumor óseo), y que han sido dados de alta del Servicio de Pediatría del Hospital de la Santa Creu i Sant Pau.
Se ha administrado el QOL-CS (Ferrel et al, 1995) en una versión adaptada de la traducción en lengua hispánica (Juárez et al, 1998). Este cuestionario consta de 41 ítems tipo Likert (1–10) que miden el bienestar de los supervivientes en los ámbitos físico, psicológico, social y espiritual.
El análisis estadístico llevado a cabo con el paquete estadístico SPSS v 11.0. incluye una comparación entre grupos diagnósticos (test de U de Mann–Whitney) y un análisis multivariante de la covarianza (Mancova) con el diagnóstico y el tipo de tratamiento como factores entre grupos.
Resultados: De los 42 supervivientes que componen la muestra, 25 son varones (59,5 %). En el momento de la evaluación la edad media es de 20,37 años, el 64,3 % de los jóvenes tienen estudios secundarios finalizados, el 57,1 % está estudiando frente al 28,6% que trabaja, y el 23,8% tiene pareja estable. El 76,2 % de la muestra son supervivientes de leucemia que han recibido TMO y con una edad media en el momento del diagnóstico de 8,03 años. El 23,8% restante son supervivientes de tumor óseo, con 14,3 años de media de edad en el momento del diagnóstico, y de los cuales el 70 % fue sometido a quimioterapia (QT) y cirugía conservadora del miembro, mientras que el 30 % recibió QT y amputación del miembro afectado. La puntuación media de CV en la muestra es de 6,58. Las puntuaciones de CV más elevadas son las que se refieren al bienestar físico en el momento actual. Se han obtenido diferencias estadísticamente significativas entre los dos grupos en la CV total según el tipo de cáncer sufrido (p = 0,01): los supervivientes de leucemia informan de una CV total, física, psicológica, social y espiritual más elevada que los supervivientes a tumores óseos. Se obtienen resultados diferenciales según el tratamiento recibido para el tumor óseo: los que han salvado el miembro obtienen niveles más elevados de CV psicológica y social que a los que se les ha sido amputado, mientras que estos últimos puntúan más alto en CV física y espiritual. El análisis multivariante de la covarianza revela una interacción significativa entre el diagnóstico y el tratamiento recibido para la CV física (p = 0,024), psicológica (p = 0,008) y espiritual (p = 0,011).
Conclusiones:1) Los jóvenes supervivientes informan de niveles moderadamente altos de bienestar. En concreto, estos niveles son más altos en lo que respecta al bienestar físico y social, en comparación con el bienestar psicológico o espiritual. 2) Los supervivientes de leucemia con TMO informan de mayor bienestar que los de tumor óseo. 3) Tanto el diagnóstico como el tipo de tratamiento recibido influyen por separado y conjuntamente en los niveles de CV comunicados por los supervivientes. 4) Las puntuaciones más elevadas en CV física y espiritual entre los diagnosticados de tumor óseo y que sufren la amputación del miembro en comparación con los que conservan el miembro pueden sugerir la presencia de factores psicológicos (p. ej., la negación del malestar) que intervienen en la respuesta de los supervivientes a la enfermedad y al tratamiento, y que están relacionados con el constructo de “resiliencia”. 5) Parece interesante poder complementar los estudios cuantitativos sobre CV con estudios cualitativos que permitan ahondar en las variables psicológicas implicadas en el bienestar subjetivo que informan los supervivientes de cáncer infanto-juvenil.
NEOPLASIAS EN PACIENTES CON ANEMIA DE FANCONI TRAS TRASPLANTE DE PROGENITORES HEMATOPOYÉTICOS: NUEVOS DESAFÍOS DE UNA VIEJA ENFERMEDADI. Elorza, C. Díaz de Heredia, T. Olivé, J. Sánchez de Toledo.
Servicio de Oncología y Hematología Pediátrica. Hospital Universitario Vall d'Hebron. Barcelona. España.
Introducción: La anemia de Fanconi (AF) es una enfermedad constitucional caracterizada por fragilidad cromosómica, insuficiencia medular progresiva y malformaciones congénitas. Los pacientes tienen riesgo de desarrollar síndrome mielodisplásico (SMD), leucemia mieloblástica aguda (LMA) y tumores sólidos, principalmente carcinomas de cabeza y cuello y ginecológicos. El riesgo estimado de presentar estas neoplasias, según diferentes estudios, es del 52 % a los 40 años y del 75 % a los 45 años. Los regímenes de quimioterapia convencional en el tratamiento de los SMD y LMA no se han demostrado eficaces, por lo que estos pacientes se consideran candidatos a trasplante de progenitores hematopoyéticos (TPH).
Material y métodos: En nuestro centro han sido sometidos a TPH 30 pacientes afectados de AF, de los cuales han sobrevivido a largo plazo 20. Presentamos 5 pacientes que han desarrollado segundas neoplasias postrasplante: 1 LMA, 1 SMD tipo anemia refractaria con exceso de blastos (AREB), 1 carcinoma epidermoide de lengua, 1 carcinoma basocelular y 1 adenocarcinoma faríngeo.
Resultados: Un total de 2 pacientes trasplantados en el contexto de un SMD al diagnóstico, desarrollaron 2 años postrasplante una hemopatía maligna: LMA y SMD. Ambos pacientes recibieron un segundo trasplante con evolución favorable, aunque el paciente que presentó el SMD desarrolló 10 años después del segundo trasplante una LMA M5, que está en fase de tratamiento. Otros 3 pacientes presentaron carcinomas 15, 19 y 20 años postrasplante a las edades respectivas de 24, 27 y 31 años. El paciente con adenocarcinoma faríngeo falleció. El paciente afectado de carcinoma basocelular evolucionó favorablemente tras exéresis quirúrgica, así como el paciente con carcinoma epidermoide de lengua, que requirió radioterapia adyuvante además de exéresis de la lesión.
Conclusiones: A pesar de que el TPH puede curar la situación de insuficiencia medular, la adquisición de aberraciones cromosómicas progresivas y la toxicidad de los tratamientos dan lugar a neoplasias que ensombrecen el pronóstico a largo plazo. El tratamiento de estas neoplasias no está bien establecido actualmente. La mejora de los resultados del TPH en estos pacientes nos permitirá conocer la evolución a largo plazo y diseñar medidas preventivas y estrategias terapéuticas adecuadas.
INFECCIÓN POR VIRUS RESPIRATORIO SINCITIAL EN EL PACIENTE ONCOHEMATOLÓGICOJ.L. Dapena1, C. Díaz de Heredia1, C. Figueras2, I. Calicó3, E. Roselló3, P. Bastida1, A. Llort1, D. Vizmanos1, T. Olivé1 y J. Sánchez de Toledo1
1Servicio de Oncología y Hematología Pediátrica. 2Unidad de Infectología Pediátrica. 3Servicio de Microbiología Clínica. Hospital Universitario Vall d'Hebron. Barcelona. España.
Introducción: Las infecciones por virus respiratorio sincitial (VRS) son una causa importante de enfermedad respiratoria en el paciente oncohematológico y, especialmente, en el paciente trasplantado y ocasionan morbimortalidad. Además, pueden ser causa de infección nosocomial. Sin embargo, su historia natural y su tratamiento todavía no están bien establecidos. La ribavirina ha sido el tratamiento antiviral más utilizado. Palivizumab, un anticuerpo monoclonal desarrollado para la profilaxis frente a VRS en los niños prematuros, podría tener un papel en el tratamiento de estos pacientes.
Material y métodos: Se han revisado las historias clínicas de los pacientes con infección respiratoria por VRS diagnosticada entre los años 1999 y 2007.
Resultados: Durante este periodo se han diagnosticado en nuestro centro 212 leucemias agudas y se han realizado 325 transplantes de progenitores hematopoyéticos. Un total de 40 pacientes de edades comprendidas entre 4 meses y 16 años presentaron clínica de infección respiratoria asociada a aislamiento de VRS. De éstos, 27 habían recibido un trasplante de progenitores hematopoyéticos, 11 presentaban leucemia aguda y 2, inmunodeficiencias congénitas. Los pacientes presentaron infección respiratoria por VRS independientemente de su edad. Pese a que la mayoría de infecciones (90 %) ocurrieron en los meses de noviembre a febrero, en 4 casos fueron más tardías. En 37 pacientes la evolución fue favorable y en 3, desfavorable. Entre enero de 1999 y noviembre de 2006 se diagnosticaron 32 pacientes positivos de VRS, de los cuales 10 recibieron tratamiento con ribavirina. En el año 2006, 3 de estos pacientes desarrollaron neumopatía con evolución fatal, por lo que a partir de entonces, con el objetivo de evitar la progresión de la infección, se inició un tratamiento precoz con la combinación de ribavirina i.v. y palivizumab. Un total de 8 pacientes positivos al VRS han recibido este tratamiento desde diciembre de 2006, con evolución favorable de su cuadro respiratorio.
Conclusiones: Los pacientes oncohematológicos pueden sufrir infecciones respiratorias por VRS, que aunque no son muy frecuentes ocasionan morbimortalidad. Estas infecciones pueden presentarse a cualquier edad, y aunque predominan en meses fríos no deben olvidarse en pacientes inmunodeprimidos fuera de su época estacional. Un tratamiento profiláctico generalizado al paciente con cáncer no parece justificado. Sin embargo, un tratamiento precoz de la infección podría tener su función en evitar el desarrollo de neumopatía grave. La combinación de ribavirina i.v. y palivizumab parece ser una alternativa eficaz en estos pacientes.
EFICACIA Y SEGURIDAD DE GENTUZUMAB OZOGAMICÍN EN NIÑOS CON LEUCEMIA AGUDA RECIDIVADA O REFRACTARIAA. Llort, P. Bastida, C. Díaz de Heredia, J.L. Dapena y J. Sánchez de Toledo
Servicio de Oncohematología Pediátrica. Hospital Vall d'Hebron. Barcelona. España.
Objetivos: Evaluar la eficacia y la seguridad del tratamiento con gentuzumab ozogamicín (Mylotarg; CMA676, GO), un anticuerpo monoclonal con unión específica al Ag CD33 de la superficie de las células leucémicas y también de células mielomonocíticas inmaduras.
Métodos: Realizar un estudio retrospectivo de 8 pacientes afectados de LAM refractarias (5) o recidivadas (3) que se trataron con GO. Los pacientes recibieron quimioterapia inicial según el protocolo Pethema LAM2002 entre diciembre de 2002 y enero de 2007. El protocolo incluye un tratamiento de inducción (daunorrubicina + AraC + etopósido) y 3 ciclos de consolidación (AraC + mitoxantrona/AraC + MAMSA/AraC + fludarabina), seguido de un trasplante de médula ósea (TMO) según los factores de riesgo. En la recaída o refractariedad todos presentaban positividad CD33 > 20% y recibieron GO en régimen de uso compasivo. La infusión fue de 2h con una dosis de 46mg/m2 en dosis única asociado a quimioterapia (citarabina + fludarabina). Los pacientes fueron evaluados según el grado de remisión, supervivencia y aparición de toxicidad usando criterios estandarizados.
Resultados: La edad media en el momento del diagnóstico fue de 7,5 años (rango 17 meses y 16 años). De ellos, 3 eran LAM recidivadas a los 9 meses, 2,5 y 3 años, respectivamente, del diagnóstico inicial y los 5 restantes eran refractarias. El GO se administró en combinación con QMT, con un intervalo máximo de 10 días. Todos recibieron una única dosis de GO, 6 pacientes de 4mg/m2y 2 de 6mg/m2. El 100% recibió premedicación con antihistamínicos y corticoides. La tolerancia al tratamiento fue buena en la mayoría. De los 8 pacientes, 3 (37 %) presentaron fiebre durante o hasta 24h posinfusión como único efecto relacionado con la infusión, que cedió con una única dosis de antitérmicos. Otros 3 pacientes presentaron cuadro de hipotensión leve que se normalizó con administración de volumen (máximo 10mg/kg). Los 2 pacientes restantes presentaron cuadro de anafilaxia que requirió una dosis de adrenalina y metilprednisolona. Ninguno de ellos presentó enfermedad venooclusiva hepática (3 niños habían recibido un TPH previo) ni otros signos de hepatotoxicidad. El 100% presentó neutropenia, anemia y plaquetopenia (III-IV) de 34 semanas. Las respuestas al GO fueron la siguientes: en 5 pacientes (62 %) se alcanzó la remisión completa; además, 3 de estos 5 recibieron TPH pos-GO. De los otros 3 pacientes, 2 mostraron una respuesta parcial y nula respuesta en el paciente restante. De ellos, 2 fallecieron por progresión de la enfermedad. La mediana de seguimiento fue de 9 meses (1–36 meses).
Conclusiones: La combinación de GO con AraC + fludarabina es bien tolerada. Los principales efectos secundarios fueron la fiebre y la mielosupresión. Con la combinación de quimioterapia + GO como tratamiento de inducción en pacientes pediátricos con AML refractaria o recidivada, se obtienen respuestas superiores al 50 %, aunque se requiere de un estudio más amplio, prospectivo y controlado para valorar correctamente su eficacia.
TROMBOCITOSIS EN LA CONSULTA DE ONCOHEMATOLOGÍA INFANTILC. Mata Fernández, M. Etxeverria Fernández, E. Cela de Julián, C. Beléndez Bieler, N. Naut Suberví, A. Cantalejo López y P. Galarón García
Servicio de Oncohematología Infantil. Hospital Gregorio Marañón. Madrid. España.
Objetivos: Evaluar las características y la etiología de la trombocitosis (TB) en niños derivados para estudio a una consulta de oncohematología infantil.
Material y métodos: Se revisan 33 niños estudiados por TB en los últimos 15 meses. Se analiza la edad en el momento del diagnóstico, el sexo, la procedencia, los antecedentes personales (AP) y familiares (AF), cifra de plaquetas por la que consultan y motivo del hallazgo, cifra máxima de plaquetas en el seguimiento, valor de Hb, VCM, HCM, cifra de leucocitos, porcentaje de neutrófilos y linfocitos en el diagnóstico, exploraciones complementarias realizadas, diagnóstico etiológico y número de consultas antes del alta. La clasificación de la TB es: leve: 500-700 × 103/mm3; moderada: 700-900 × 103/mm3; grave: 900-1.000 × 103/mm3; extrema > 1.000 × 103/mm3.
Resultados: Rango de edad: desde 15 días hasta 12 años (46% menores de 24 meses); 19 varones y 14 mujeres. Procedencia: el 55% del centro de salud, el 16% del servicio de urgencias y el 29% de otros servicios del hospital. AP: 1 con síndrome de Noonan y 1 con betatalasemia minor. AF: sin interés. Cifra media de plaquetas en la primera consulta: 669.000; la mínima era de de 412.000 y en 4 pacientes (12%) era superior a 1.000.000. Motivo de la analítica: habitual (4 pacientes), preoperatorio (2), infección (9: 1 bronquiolitis, 1 neumonía con empiema, 3 infecciones bacterianas graves y 4 víricas). En el seguimiento llegaron a cifras extremas 8 niños (24%). El 90% de las TB leves no pasaron de cifras moderadas. De los 8 casos de TB extrema, 1 era reactiva a anemia neonatal, 4 reactivas por ferropenia, 2 eran infecciones de vías respiratorias bajas (IVRB) y 1, pielonefritis. Presentaban leucocitosis al diagnóstico 12 (36%), con linfocitosis 6 y neutrofilia 2. El número medio de visitas hasta el estudio fue de 5; sólo el 46% ha sido dado de alta. Exploraciones complementarias: metabolismo del hierro en el 87 % (29), detectándose ferropenia en el 55 % de éstos (16), de los cuáles sólo 1 (betatalasemia) presentaba VCM y HCM por debajo de −2 DE; estudio de inmunoglobulinas al 27%, serologías y hormonas tiroideas al 21 %, autoinmunidad al 6 %, eco abdominal al 24 %, y otros en menor número. El diagnóstico etiológico de nuestros casos ha sido: 12TB reactivas a infecciones (36%), 8 por ferropenia (24%), 7 etiología mixta (21%) (reactiva y ferropenia), 1 betatalasemia y ferropenia asociadas (3%), 2 secundarias a anemia neonatal (6 %) y 3 en estudio (9 %) (también con características de secundarias).
Conclusiones: Las TB son un hallazgo frecuente en la infancia, por lo que suponen un motivo frecuente de consulta en los servicios de oncohematología pediátrica. Proceden principalmente del centro de salud o del propio hospital, y el hallazgo es mayoritariamente casual o en el contexto de un cuadro infeccioso. La TB primaria es extremadamente rara en la infancia. Se clasifica en TB familiar y TB clonal (trombocitemia esencial y síndromes mieloproliferativos). Nuestro estudio corrobora que a diferencia de las primarias, las TB secundarias se caracterizan por predominar en la primera década de la vida, especialmente en los primeros 2 años, ser transitorias, con cifras leves o moderadas de TB, frecuente fiebre y signos de infección y generalmente no cursan con esplenomegalia. Suelen ser TB leves y en bajo porcentaje extremas (éstas son un hallazgo frecuente en infecciones de vías respiratorias bajas en las que marcan gravedad y en la enfermedad de Kawasaki). Las alteraciones plaquetarias y de la coagulación son frecuentes en el síndrome de Noonan. Además, las principales causas de TB secundaria en la infancia son: infecciones o inflamaciones agudas o crónicas, ferropenia, pérdida aguda de sangre y menos frecuentes: anemia hemolítica, anesplenia y rebote posesplenectomía.
PÓSTERSLEUCEMIA LINFOBLÁSTICA T: ¿RECIDIVA MUY TARDÍA O SEGUNDA NEOPLASIA? CASO CLÍNICOA. Llort, P. Bastida, C. Díaz de Heredia, I. Elorza, J.L. Dapena y J. Sánchez de Toledo
Servicio de Oncohematología Pediátrica. Hospital Vall d'Hebron. Barcelona. España.
Objetivos: Actualmente los niños afectados de leucemia aguda linfoblástica (LAL) tienen unas posibilidades de curación por encima del 80 %. La aparición de recidivas muy tardías después del diagnóstico inicial son muy escasas y plantean la necesidad del diagnóstico diferencial con segundas neoplasias.
Método: Descripción y análisis de una niña diagnosticada de leucemia aguda linfoblástica de fenotipo B común a la edad de 2 años y que presenta, 9 años más tarde, una nueva infiltración medular. Estudio de la bibliografía al respecto.
Resultados: A los 2 años (1997) fue diagnosticada de LAL B común, a raíz de un síndrome constitucional, adenopatías y hepatoesplenomegalia. La médula ósea (MO) demostró infiltración masiva por blastos L1, con el siguiente inmunofenotipaje: marcadores B = CD19 47%, CD10 33%, CD10-CD19 33%, CD22 46 %, marcadores T = negativos y otros marcadores = CD45 92%, CD34 29%, TdT positivo. La citogenética de la MO fue normal. Se aplicó quimioterapia según protocolo Pethema-89 con buena respuesta y finalizó el tratamiento a los 2. años. A los 11 (9 años después del diagnóstico inicial) presentó de nuevo adenopatías laterocervicales y supraclaviculares. En MO se observó una infiltración del 71% de células blásticas de morfología L1. En la citometría de flujo de MO había marcadores positivos T: CD3, TDT, CD2, CD5, CD11b, etiquetándola de leucemia linfoblástica T (T-II EGIL). Además, la citogenética de la MO fue 46 XX, 5q-(3)/46 XX,5q-, 7q-, 12p+(2). Mediante FISH el 66,5% presentaban pérdida de 5q31. Se inició tratamiento según protocolo de alto riesgo (LAL-AR-2005) con mala respuesta el día 14 del mismo. Recibió quimioterapia de consolidación y fue sometida a trasplante de precursores hematopoyéticos (TPH) de donante no emparentado. Actualmente, permanece en remisión completa a los 6 meses pos-TPH.
Conclusiones: Este caso plantea la hipótesis del diagnóstico de una recidiva muy tardía de la leucemia inicial pero con selección de la clona menor, detectada o no en el diagnóstico inicial, pero con una expansión más lenta y más resistente a la quimioterapia (linfoblastos T) o bien un cambio de las características inmunológicas de las células leucémicas o bien que se trate de una segunda neoplasia. Un dato a favor de recidiva es que la delección 5q-, se asocia al síndrome mielodisplásico secundario a quimioterapia y leucemias agudas mieloblásticas.
RÉGIMEN HIPERTRANSFUSIONAL EN UN ADOLESCENTE CON TALASEMIA INTERMEDIAE. Aleo Luján y C. Gil López
Unidad de Oncohematología Pediátrica. Hospital Clínico San Carlos. Madrid. España.
Introducción: Paciente varón que fue diagnosticado a los 3 años de edad de talasemia intermedia: betatalasemia heterocigota y hemoglobina Lepore.
Material y métodos: Desde el diagnóstico y hasta la edad de 13,6 años recibió un total de 27 transfusiones de concentrado de hematíes coincidiendo con procesos intercurrentes entre los que destacamos:
- –
Apendicitis aguda a los 5,5 años de edad.
- –
Esplenectomía y colecistectomía a los 8,6 años de edad.
- –
Sepsis por yersinia enterocolítica y nefropatía por IgA a los 10,3 años de edad.
A los 9,3 años comenzó quelación de hierro con desferroxiamina subcutánea (25mg/kg, 5 días a la semana) con una ferritina de 1.020ng/ml, se añadió al tratamiento vitamina C y E.
A los 12 años se comenzó a observar un enlentecimiento de las curvas de crecimiento con un retraso de 2 años en la edad ósea y sin inicio del desarrollo puberal. Se realizó estudio hormonal, en el que se apreció una hiposecreción de GH y de somatomedina C. La situación de estancamiento en el desarrollo puberal y crecimiento se prolonga hasta los 13 años con igual situación endocrinológica y con valores de ferritina de 821ng/ml por lo que se decide iniciar régimen hipertransfusional. Realizamos transfusiones de concentrado de hematíes (2 unidades) cada 3 o 4 semanas intentando mantener las cifras de hemoglobina entre 10 y 12g/dl continuando con la misma terapéutica de quelación hasta los 15,9 años, cuando se suspende la quelación con desferroxiamina subcutánea y entra a participar en el ensayo CICL 670A2409 con el quelante oral del hierro ICL670 (EXJADE). Con el régimen hipertransfusional, partimos de una talla de 142cm para llegar en el momento actual a una talla de 170cm, habiendo alcanzado su talla genética. Las velocidades de crecimiento se incrementaron ya desde los primeros meses de inicio de la terapia con niveles de 10cm/año, presentó desarrollo puberal alcanzando un volumen testicular adulto a los 2,5 años de iniciar este régimen.
Desde el punto de vista de la sobrecarga férrica, los primeros 2 años de terapia hipertransfusional incrementaron su concentración de ferritina hasta 1.871ng/ml con el tratamiento con desferroxiamina. Con la terapia de quelación actual la ferritina se mantenía en concentraciones similares a las del inicio, durante la primera parte del estudio. Posteriormente, aumentó de peso y se elevó la dosis recibida de quelante. Sufrió un segundo incremento por una elevación ligera de las cifras de Ferritina llegando a una dosis de 25mg/kg de EXJADE, a pesar de lo cual no hemos conseguido rebajar las cifras de ferritina, que se encuentran actualmente en 2.500ng/ml.
Conclusiones: Presentamos el caso de un paciente que había seguido el curso clínico esperado en una talasemia intermedia, con transfusiones coincidiendo con procesos intercurrentes, pero que, llegado el momento de realizar la pubertad, ésta no se presentó. Esto podría deberse a sobrecarga férrica, pero sus cifras de ferritina no parecían indicar esta causa, por lo que decidimos probar el régimen hipertransfusional, que ha sido determinante para conseguir un crecimiento y desarrollo normales. Además, ha tenido la oportunidad de participar en el estudio con EXJADE, aunque los resultados en este paciente no han sido muy alentadores, probablemente por necesitar una dosis más elevada.
REVISIÓN DE NUESTRA EXPERIENCIA CON LOS CASOS DE PÚRPURA TROMBOCITOPÉNICA IDIOPÁTICAC. Ruiz Ledesma, C. López Calero, F. Vela Casas y J. Sánchez Calero
Hospital Universitario Virgen Macarena. Sevilla. España.
Objetivos: Exponer nuestra experiencia sobre la púrpura trombocitopénica idiopática (PTI) diagnosticada en niños en nuestro servicio desde el año 1996 hasta 2005.
Material y métodos: Se estudian de forma retrospectiva los casos de PTI en dichos años, atendiendo a la edad, sexo, clínica, complicaciones de la enfermedad, tratamientos realizados y respuesta, cronicidad o no de la enfermedad y duración del seguimiento. Consideramos criterios de inclusión: plaquetopenia < 100.000/μl, ausencia de infecciones, medicamentos y hallazgos clínicos o de laboratorio (hepatoesplenomegalia, anemia, etc.) que nos hagan pensar en otras enfermedades.
Resultados: En dicha década se diagnostican en nuestro servicio 22 niños, con un predominio femenino sobre el masculino (16:6) y una edad media de 5 años. Un total de 7 casos fueron PTI crónica y el resto, agudas. En 10 años tenían como antecedente algún proceso infeccioso semanas antes y 1 tenía un familiar afectado. En 1 caso se pudo demostrar la presencia de anticuerpos antiplaquetarios en sangre. Todos los casos presentaron manifestaciones cutáneas (petequias, equimosis, hematomas) y, además, en 3 casos gingivorragia, en 2 epistaxis y en 1 hematuria. En el momento del diagnóstico, 13 niños tenían plaquetopenia < 20.000/μl.
Tratamiento: 2 casos no precisaron tratamiento, en 8 se usaron únicamente corticoides (respuesta completa [RC] en 7 niños), en 3 sólo inmunoglobulinas (RC en 2), corticoides e inmunoglobulinas en 7 niños (RC en 4). En 2 niños con PTI crónica fue necesario la esplenoctomía con posterior remisión completa en ambos casos. La duración media del seguimiento fue de más de 2 años en la PTI crónica y aproximadamente 1 año para la PTI aguda.
Conclusiones: Aunque la etiopatogenia más aceptada en la actualidad es la destrucción plaquetaria por mecanismo autoinmune, la ausencia de anticuerpos antiplaquetarios no excluye el diagnóstico (sólo demostrable en uno de nuestros casos). Como se describe en la literatura médica, en nuestros niños es frecuente el antecedente infeccioso y las manifestaciones cutáneas sin otros signos de alarma (mal estado general, adenopatías, etc.). Al igual que en otros protocolos, nuestro tratamiento dependió fundamentalmente de las cifras plaquetarias, y el tratamiento estándar fueron los corticoides en dosis elevadas por vía oral y las gammaglobulina i.v. En los casos de PTI crónica que no responden a estos tratamientos, puede ser necesario recurrir a la esplenectomía.
QUELACIÓN CON DEFERIPRONA EN 3 PACIENTES ADOLESCENTES AFECTADOS DE BETATALASEMIA MAYORC. Villafruela Álvarez1, R. López almaraz1, J. Rodríguez Luis1 y B.J. González González2
1Servicio de Pediatría (Oncohematología pediátrica). 2Servicio de Hematología y Hemoterapia. Hospital Universitario de Canarias. Tenerife. España.
Objetivo: Presentar la evolución, eficacia y seguimiento de las posibles complicaciones de 3 pacientes de raza hindú afectados de betatalasemia mayor en tratamiento quelante con deferiprona.
Pacientes y métodos: Seguimos durante 4 años a 3 hermanos de raza hindú afectados de betatalasemia mayor (2 varones y 1 mujer), en la consulta externa de hematooncología pediátrica y en la de hematología de adultos. Todos recibían transfusiones de hematíes con una media mensual desde su diagnóstico y quelación continua con desferroxiamina subcutánea en bomba de infusión. Hace 4 años, se decide cambiar la quelación con desferroxiamina, por la oral con deferiprona con el objeto de mejorar la calidad de vida y la adherencia al tratamiento. Evaluamos la eficacia, tolerancia y evolución de su enfermedad tras la introducción de la deferiprona.
Resultados: La introducción de deferiprona se realizó en todos los casos en la adolescencia, comprobándose concentraciones de ferritina estables (de 200 a 250mg/dl de media) con un descenso significativo respecto a los previos (150 a 250mg/dl), perfil hepático conservado, ausencia de acumulación férrica y de fibrosis en tejido cardíaco (controles ecocardiográficos) y hepático (control de resonancia magnética) en los 3 pacientes. Ninguno ha presentado efectos adversos secundarios con esta medicación, y han permanecido la serie blanca, el perfil hepático y la función gastrointestinal conservados. Tanto la familia como los pacientes valoran el cambio como una mejoría importante en su calidad de vida y en la adherencia al tratamiento.
Conclusión: La deferiprona ha sido una alternativa muy eficaz y bien aceptada con respecto al tratamiento quelante subcutáneo, en pacientes adolescentes afectados de betatalasemia mayor.
AFECTACIÓN NEUROLÓGICA POR EL VIRUS DE LA VARICELA-ZÓSTER EN UN PACIENTE DE 16 MESES DE EDAD SOMETIDA A TRASPLANTE DE SANGRE DE CORDÓN UMBILICALD. Vizmanos1, J. Dapena1, C. Díaz de Heredia1, T. Olivé1, C. Figueras2, N. Mayolas3 y J. Sánchez de Toledo1
1Servicio de Oncohematología Pediátrica. 2Servicio de Infectología Pediátrica. 3Servicio de Radiología Pediátrica. Hospital Materno-Infantil Vall d'Hebron. Barcelona. España.
Introducción: La reactivación del virus de la varicela-zóster (VVZ) es un proceso común que puede acarrear complicaciones graves en los pacientes sometidos a trasplante de progenitores hemopoyéticos (TPH). La afectación del sistema nervioso central (SNC) por este virus es, no obstante, poco frecuente en estos pacientes, y está poco descrita en la literatura.
Caso clínico: Presentamos el caso de una niña de 16 meses afectada de leucemia aguda mielobástica M5 en segunda remisión, con antecedentes de varicela a los 5 meses de edad. En el curso de los diferentes tratamientos quimioterápicos, se reactivó en diversas ocasiones la infección cutánea y respondió bien al tratamiento con valaciclovir. A su ingreso en la unidad de TPH presentaba mínimas lesiones costrosas en el cuero cabelludo; recibió profilaxis secundaria con aciclovir intravenoso en 1.500mg/m2/día. Se sometió a un trasplante de sangre de cordón umbilical. El día +39 requirió tratamiento con corticoides i.v. en 2mg/kg/día por EICH cutáneo grado 3, por lo que se aumentó la dosis a 5mg/kg/día el día +43 por empeoramiento del cuadro. Tras 5 días, la paciente presentó una polineuropatía de pares craneales (II y III) con neuritis óptica izquierda. Se descartó la recidiva leucémica neuromeníngea y la PCR a VVZ fue positiva en líquido cefalorraquídeo (LCR). Se inició tratamiento con foscarnet i.v. y gammaglobulinas i.v. semanales manteniéndose el aciclovir en 1.500mg/m2/día, y se disminuyó la dosis de metilprednisolona hasta suspenderse en el día +78. Presentó como complicación una hidrocefalia arreabsortiva que requirió la colocación de un drenaje ventricular externo en el día +71, que se cambió por una válvula de derivación ventriculoperitoneal en el día +95, tras haberse normalizado la citoquimia del LCR y negativizado la PCR del VVZ en LCR. La paciente presentó una lenta mejoría hasta la resolución de la afectación bilateral del III par craneal. Presentó como secuela de la neuritis y retinitis bilateral, una amaurosis bilateral persistente en el día +131 y un deterioro cognitivo. Recibió tratamiento antivírico durante más de 3 meses.
Conclusión: En pacientes sometidos a TPH que presentan signos y síntomas neurológicos multifocales, aunque poco es frecuente, habrá que descartar siempre la infección del SNC por VVZ.
HEPATITIS TÓXICA EN RELACIÓN CON LA ADMINISTRACIÓN DE METOTREXATO (MTX) POR VÍA INTRATECALC. Herrero, M. Martínez, Ll. Hernández, F. Almazán, W. Coroleu y G. Javier
Servicio de Pediatría. Hospital Universitario Germans Trias i Pujol. Badalona. Barcelona. España.
Objetivos: El MTX es un antimetabolito antineoplásico de mecanismo de acción y toxicidad bien establecidos. Es conocida la hepatotoxicidad derivada de su uso por vía sistémica, pero no hemos encontrado en la literatura médica casos de hepatotoxicidad secundaria a su administración exclusiva por vía intratecal.
Material y métodos: Describimos el caso de un recién nacido a término que ingresa por presentar hepatosplenomegalia, estrés respiratorio y nódulos cutáneos. Se diagnostica de leucemia aguda congénita. Se inicia tratamiento según protocolo SHOP LAL Lactantes 02 con buena respuesta hematológica con desaparición de blastos en sangre periférica y médula ósea el día +14. Se le administra triple terapia intratecal (MTX 6mg, arabinosido de citosina 16mg, hidrocortisona 8mg) el día +1 y +7 según protocolo.
Resultados: El paciente presenta ictericia progresiva, que se hace manifiesta a las 72h de la administración de la segunda dosis de MTX intratecal (día +7). El estudio analítico muestra signos de hepatitis no colestática (GOT 2.813 U/l, bilirrubina total 18,8mg/dl, bilirrubina directa 1,3mg/dl) sin signos clínicos ni de laboratorio de hemólisis. Tras descartarse posibles etiologías infecciosas o medicamentosas, se realiza determinación de concentraciones séricas de MTX, que sonde 0,12 micromol/l. Se consigue recuperar suero correspondiente a las 24h posadministración de MTX en que las concentraciones del mismo eran de 0,5 micromol/l. Resolución espontánea posterior del cuadro de hepatitis, con concentraciones a las 90h de MTX de 0,05 micromol/l. El paciente fallece a los 20 días de vida como consecuencia de un cuadro séptico con fallo multiorgánico.
Conclusiones: El mal pronóstico de la leucemia congénita viene determinado tanto por las características especiales de la enfermedad como por la inmadurez y el difícil tratamiento clínico del paciente. No hemos encontrado descripciones de hepatotoxicidad sistémica del MTX secundarias a su administración exclusiva por vía intratecal.
El caso clínico descrito hace pensar en la necesidad de estudiar las concentraciones séricas de MTX y de la curva de eliminación del mismo en recién nacidos y lactantes pequeños y la eventual utilización de un rescate con ácido folínico en función de las concentraciones séricas obtenidas. Las características de inmadurez de la barrera hematoencefálica del neonato posibilitarían un mayor paso de este fármaco a la circulación sistémica con la consiguiente elevación de las concentraciones séricas y una posible toxicidad secundaria al mismo.
LEUCEMIA AGUDA BILINEAL DE PRESENTACIÓN CONGÉNITAF. Almazán1, C. Herrero1, J. Junca1, I. Granada1, M. Forns M2, P. Bastida2 y G. Javier2
1Servicio de Pediatría. Hospital Universitario Germans Trias i Pujol. Badalona. Barcelona. 2Servicio de Hematología. Hospital Universitario Materno-Infantil Vall d'Hebron. Barcelona. España.
Objetivos: La leucemia aguda congénita es una entidad que representa solamente el 1 % del total de las leucemias en la infancia. En muchos casos estas leucemias son bifenotípicas. Sin embargo, las formas bilineales son de presentación extremadamente infrecuente.
Material y métodos: Presentamos 2 casos de leucemia congénita bilineal diagnosticados mediante inmunofenotipo y biología molecular.
Resultados: Caso 1: Recién nacido a término que ingresa por estrés respiratorio, hepatoesplenomegalia y nódulos cutáneos sugestivos de leucemia cutis. El hemograma muestra leucocitosis (95 × 109/l) con el 80% de blastos en los que morfológicamente se observa una doble población (linfoide y monocitoide). El inmunofenotipo confirma dos poblaciones diferenciadas: una blastosis con marcadores de línea monocítica y una población linfoide B que sólo expresa CD19 y CD38. El estudio citogenético y de biología molecular muestra t (4;11), monosomias 8 y 9, delección 11p11.2 y dos cromosomas marcadores, reordenamiento MLL+ y clonalidad para la IgH. Se trata con protocolo LAL-lactantes SHOP-02 y se consigue la remisión hematológica medular en el día +14. No obstante, la aparición de diversas complicaciones (síndrome de lisis tumoral pese a la correcta profilaxis del mismo, sepsis por Klebsiella, toxicidad hepática por metotrexato, síndrome de secreción inadecuada de ADH), hacen que el paciente fallezca a los 20 días de vida. Caso 2: Paciente de 3 meses de vida sin antecedentes que ingresa para estudio de masa abdominal. Por ecografía se comprueba que corresponde a esplenomegalia y los exámenes complementarios muestran hiperleucocitosis (470 × 109/l) con el 99 % de blastos. El estudio morfológico e inmunofenotípico revelan una leucemia con dos poblaciones diferentes, una línea predominante (el 82 % de blastos) con inmunofenotipo pro-B y una minoritaria (13 %) con diferenciación monocitaria. La citogenética muestra t (4;11) y por FISH se confirma el reordenamiento MLL. El paciente presenta signos de CID incipiente, por lo que es trasladado a unidad de cuidados intensivos. Se practica exanguinotransfusión dada la gran leucocitosis y simultáneamente se inicia tratamiento según protocolo mixto (PETHEMA LAL AR 2005/Interfant 99), al que presenta una buena respuesta (AMO día +14: ausencia de blastos).
Conclusiones: El mal pronóstico de la leucemia congénita es debido no sólo a sus propias características diferenciales, sino a la dificultad del tratamiento clínico del paciente.
De acuerdo con los recientes avances referentes a la base molecular de la leucemia, es razonable pensar que cualquier leucemia detectada en los primeros meses de vida pueda ser de origen congénito. Es extremadamente infrecuente el diagnóstico de leucemia congénita bilineal, aunque es posible que la afectación de elementos muy inmaduros del sistema hematopoyético, más frecuente a estas edades, pueda condicionar la afectación de más de una línea celular.
ENFERMEDAD DE ERDHEIM-CHESTER: UNA RAREZA EN LA INFANCIAM. García Bernal1, B. Pineda Prats B2, S. Brió Sanagustín2, M. Torrent Español2, N. Pardo García2, E. Turón Viñas2 e I. Badell Serra2
1Servicio de Pediatría. Hospital Mutua de Terrassa. 2Unidad de Hematooncología. Servicio de Pediatría. Hospital de la Santa Creu i Sant Pau. Barcelona. España.
Introducción: La enfermedad de Erdheim-Chester (ECD) es una histiocitosis no de Langerhans de muy rara presentación pediátrica. Existe infiltración tisular por histiocitos que son FXIII (+), CD68 (+), CD1A (−) y S-100 (+/−). Afecta con mayor frecuencia a las diáfisis de los huesos largos y el sistema nervioso central, especialmente a la región maxilar-orbitaria. El pronóstico es en general infausto, con curso indolente y escasa respuesta a la corticoterapia, así como a los citostáticos e immunomoduladores. Se han comunicado respuestas a interferón α y un caso de autotrasplante de médula ósea que logró la remisión. La mortalidad es del 60 % y se debe, sobre todo, a las complicaciones cardiopulmonares y renales de esta enfermedad.
Caso clínico: Niña de 18 meses con dolor en muslo izquierdo (I) y fiebre, orientado como osteomielitis por gammagrafía con Tc99 hipercaptante, por lo que recibe antibioticoterapia intravenosa. Los hemocultivos y cultivos óseos son negativos. Durante el ingreso presenta tumefacción en surco nasogeniano derecho, cuya biopsia es compatible con histiocitosis de células de Langerhans, por lo que se inicia tratamiento según protocolo SEOP con prednisona y vinblastina. Mejora parcialmente, pero aparece tumefacción en tibia distal I. Se remite a nuestro centro para completar el estudio. Entre las exploraciones realizadas destaca: aumento de VSG y ferritina, mielograma normal, radiografías simples de fémur y tibia I con reacción perióstica y biopsia de la lesión tibial poco concluyente. Se añade tratamiento con etopósido y se observa mejoría clínica. A los 2 meses presenta fiebre y tumefacción en el pie I al retirarse corticoterapia. Revalorada la biopsia inicial de la lesión maxilar (negatividad CD1A, positividad difusa de S-100 en tejido con escaso material celular) se orienta como probable osteítis crónica multifocal recurrente. Se reinicia tratamiento con prednisona y naproxeno, que producen mejoría de la clínica. Durante los 2 años siguientes sufre múltiples brotes de tumefacción ósea en diversas localizaciones, asociados a un aumento de reactantes de fase aguda y/o fiebre, por lo que se ensaya tratamiento con metotrexato, pamidronato (por osteoporosis asociada), ciclosporina, micofenolato y anti-TNF. A los 4 años, coincidiendo con la disminución de los corticoides, se objetiva protusión ocular bilateral que produce queratopatía de exposición, úlceras corneales y alteración de la visión. La RM craneal muestra masa de partes blandas sellares y orbitarias que afectan a la musculatura extrínseca ocular y nervios ópticos. En la biopsia de la lesión orbitaria se observa fibrosis orbitaria grave que engloba histiocitos CD68+, CD1a (−) y S-100 (−), compatibles con ECD. Tras recibir tratamiento con cladribina (4 ciclos) que no produce respuesta, se administra interferón α subcutáneo 3 veces/semana, con dosis que oscilan entre 4,5 y 9 × 106 UI/dosis, con lo que se obtiene una mejoría clínica mantenida y la práctica desaparición de los síntomas, con la consiguiente disminución de la corticoterapia. Actualmente recibe interferon α pegilado subcutáneo 40μg 1 vez/semana y únicamente presenta leve hipertensión arterial y cefaleas atribuidas a dicho tratamiento.
Comentarios: La ECD es extremadamente rara en la edad pediátrica y su presentación clínica plantea el diagnóstico diferencial con infecciones óseas, procesos reumáticos y otras patologías inflamatorias. La afectación ocular y el curso crónico, así como la histología, ayudan al diagnóstico. El tratamiento con interferón α es una de las pocas opciones terapéuticas eficaces.
ENFERMEDAD DE CÉLULAS FALCIFORMES: UN RETO EN HEMATOLOGÍA PEDIÁTRICAI. Elorza, A. Llort, C. Díaz de Heredia, J.L. Dapena y J. Sánchez de Toledo
Servicio de Oncología y Hematología Pediátrica. Hospital Universitario Vall d'Hebron. Barcelona. España.
Introducción: La drepanocitosis es una anemia hemolítica crónica que causa lesión progresiva de los órganos, interrumpida por episodios agudos.
Material y métodos: Presentamos un estudio retrospectivo de los pacientes con enfermedad drepanocítica (Hb SS: 25, Hb Sβtal: 3, Hb SC: 1) controlados en nuestro centro entre los años 1985 y 2006. Se han revisado 29 pacientes con edades comprendidas entre 0 y 20 años. El motivo de diagnóstico fue en el 43 % por antecedentes familiares, el 28 % por anemia y el 28 % por crisis vasooclusivas (CVO) de diferentes intensidades y localizaciones. El país de origen de los progenitores fue en el 70 % del África subsahariana (Gambia, Guinea y Senegal). Todos los pacientes han recibido vacunación y antibioticoterapia profiláctica. A lo largo de su evolución el 82% de los pacientes han presentado episodios agudos y el 28 % sufre lesión orgánica crónica. Ninguno ha presentado complicaciones durante los primeros 6 meses de vida. Las CVO son más frecuentes y graves entre las edades de 1 y 5 años, cuando se registran el 55% del total. Durante esta época 5 pacientes presentaron secuestro esplénico, 3 síndrome torácico y 2 iniciaron con accidentes cerebrovasculares. Asimismo, 3 pacientes presentaron neumonía, 1 osteomielitis y 1 meningitis bacteriana. Entre los 6 y 10 años se presentaron el 16% de las CVO. A partir de los 10 años son frecuentes las CVO óseas (5 pacientes) y los pacientes muestran ya afectación orgánica como nefropatía (2), retinopatía (3) y neumopatía (2). Aunque las complicaciones infecciosas son más frecuentes los primeros años de vida también se han detectado posteriormente osteomielitis y neumonía. Un total de 16 pacientes han recibido tratamiento con hidroxiurea en dosis de entre 15 y 30mg/kg/día, con buena respuesta y tolerancia en el 75 % de los casos. Otros 2 pacientes con afectación neurológica grave siguen régimen hipertransfusional y quelación adecuada. Se realizó trasplante de progenitores hematopoyéticos de un hermano HLA-idéntico en 2 pacientes con evolución satisfactoria en ambos. Los pacientes con drepanocitosis presentan múltiples y en ocasiones graves complicaciones.
Conclusión: La hidroxiurea parece ser un fármaco eficaz y seguro en el tratamiento de estos pacientes. Es imprescindible el diagnóstico precoz de esta enfermedad para intentar evitar los episodios agudos y retrasar el daño orgánico, realizando un seguimiento estricto dentro de un equipo multidisciplinar.
ANEMIA HEMOLÍTICA POR HIPOFOSFATEMIA GRAVE: PRESENTACIÓN POCO FRECUENTE DE UNA ENFERMEDAD DE WILSONM. Villa Alcázar1, M. Pacheco Cumani1, E. Arrontes Caballero2 y B. López-Ibor Aliño1
1Hospital San Rafael. 2Hospital Madrid Montepríncipe. Madrid. España.
Objetivo: Presentamos el caso de un paciente que empieza con una anemia hemolítica por hipofosfatemia grave como forma de presentación de una enfermedad de Wilson.
Caso clínico: Niño de 9 años, previamente sano que refiere ictericia, palidez y coluria de dos días de evolución. Una semana antes ha tenido un catarro de vías altas y un mes antes GEA autolimitada. No ha tomado medicación, no refiere ingestión de setas, contacto con pesticidas ni pegamento. Ha tocado una serpiente que no le ha mordido. Exploración física: buen estado general, palidez e ictericia III/IV de piel y mucosas, soplo sistólico II/IV, hepatomegalia de 2cm y esplenomegalia de 3cm. El resto de la exploración es normal. Evolución y tratamiento: al ingresar se realiza AS, en la que se evidencia Hb de 5g/dl con test de Coombs negativo y reticulocitos 34 × 1.000; alteración de la función hepática (con serologías de virus hepatotropos negativas) y una hipofosfatemia grave. La hemólisis cede con la corrección de la hipofosfatemia.
Se realiza: 1) estudio de causas de anemia hemolítica no inmune: PK y G-6-PD normales y extensión de SP que descarta anomalías morfológicas del hematíe, no se observan blastos. 2) Se descarta proceso linfoproliferativo asociado (Rx de tórax y ecografía abdominal normales excepto hepatoesplenomegalia homogéneas). 3) Se realiza estudio de función renal objetivándose una tubulopatía Fanconi-like sin glucosuria. Se descartan raquitismo, hipoparatiroidismo y aminoacidopatías como causas primarias de la hipofosfatemia.
La asociación de la afectación hepática con tubulopatía lleva al diagnóstico de sospecha de enfermedad de Wilson, que se confirma con determinación de las concentraciones de ceruloplasmina y cobre en sangre y orina y por la presencia de anillo de Kayser-Fleischer en examen con lámpara de hendidura. Se remite al paciente a un centro de hepatología pediátrica.
Conclusión: La enfermedad de Wilson es una alteración hereditaria del transporte del cobre en el organismo. Los síntomas principales son secundarios a la afectación hepática y/o neurológica. En algunos pacientes puede producirse una hemólisis aguda intravascular secundaria al daño oxidativo de los eritrocitos por el exceso de cobre. Menos frecuente es una anemia hemolítica secundaria a hipofosfatemia grave por tubulopatía Fanconi-like. En pacientes con anemia hemolítica Coombs negativo de etiología no filiada debe sospecharse una alteración metabólica como la enfermedad de Wilson.
LINFOHISTIOCITOSIS HEMOFAGOCÍTICA CON DEFECTO DE MUNC 13–4 DESENCADENADA POR PARVOVIRUS B19I. Elorza, C. Díaz de Heredia, J.L. Dapena, T. Olivé, D. Vizmanos y J. Sánchez de Toledo
Servicio de Oncología y Hematología Pediátrica. Hospital Universitario Vall d'Hebron. Barcelona. España.
Introducción: La linfohistiocitosis hemofagocítica (HLH) es una enfermedad rara caracterizada por la proliferación y activación no maligna de histiocitos y linfocitos T citóxicos, debida a una respuesta inmune anormal y exagerada. Aparece sobre la base de diversas condiciones que dan lugar a una misma vía común y a un estado de hiperinflamación letal para el paciente. El antecedente o la asociación a infección no permite diferenciar a los pacientes con HLH primaria y defecto intrínseco de la regulación inmune de los pacientes con HLH secundaria a infección, dado que el agente infeccioso desencadena frecuentemente en ambos, la clínica y alteraciones características de esta enfermedad. Las formas primarias se diagnostican mayoritariamente en niños menores de 2 años, y el diagnóstico fuera de este período es excepcional.
Caso clínico: Presentamos un paciente de 14 años con aplasia medular adquirida grave. En el estudio previo al trasplante de progenitores hematopoyéticos (TPH) de su hermana HLA-idéntica se objetivó primoinfección por parvovirus B19 (IgM+). Se confirmó mediante técnica de PCR la existencia del virus tanto en sangre periférica como en médula ósea. Se inició tratamiento con gammaglobulina y soporte transfusional. La médula ósea era hipoplásica, sin objetivarse otros hallazgos patológicos. El cariotipo y el estudio de fragilidad cromosómica fueron normales, y en el estudio de subpoblaciones linfocitarias destacaba una disminución de la función y el número de células natural killer (NK). Tras 15 días de tratamiento con gammaglobulina, la PCR a parvovirus permaneció positiva y apareció un exantema generalizado y fiebre alta. La fiebre persistió a pesar del tratamiento antibiótico de amplio espectro, sin objetivarse microorganismo alguno en los cultivos realizados y con las pruebas de imagen normales. El estado general del paciente empeoró progresivamente con fiebre alta, esplenomegalia y pancitopenia grave. El estudio de la medula ósea mostró entonces múltiples imágenes de hemofagocitosis. Se estableció el diagnóstico de síndrome hemofagocítico (SHF) y se inició tratamiento según protocolo HLH-2004 con ciclosporina, etopósido y dexametasona, con estabilización clínica y negativización de la PCR a parvovirus B19. Por persistencia de actividad del HLH recibió un TPH familiar idéntico tras acondicionamiento con busulfán, etopósido y ciclofosfamida. A la semana del TPH falleció por hemorragia cerebral masiva.
Conclusiones: En el estudio molecular se detectó una mutación en el gen UNC13D (17q25). Esta alteración se observa en el 25-30% de los casos de HLH tipados molecularmente. La proteína codificada por este gen, Munc 13–4, es esencial en los primeros pasos del procesamiento de lisosomas citolíticos en los linfocitos T citotóxicos. Su deficiencia origina una alteración de la exocitosis de los denominados gránulos líticos, con la consecuente alteración de la secreción de perforina y enzimas incluidas en ellos. En estos pacientes las infecciones víricas causan una respuesta inmune anormal, hipercitoquinemia generalizada y daño tisular indiscriminado. La evolución es fatal en todos los casos, por lo que es vital sospechar la HLH e iniciar el tratamiento lo más precozmente posible. En la actualidad se consigue una supervivencia del 50 %. A pesar de que el 90 % de las formas primarias se diagnostican en menores de 2 años es aconsejable realizar el estudio molecular en todos los pacientes.
HEMOGLOBINA INESTABLE “NEWCASTLE” (BETA92HIS > PRO). PRIMERA DESCRIPCIÓN EN UNA FAMILIA ESPAÑOLAJ. Sans Tojo1, M.T. Coll Sibina1, M. Mañú Pereira2, J.L. Vives Corrons2, J. Bosch1 y M. Catala1
1Servicio de Pediatría. Hospital General de Granollers. 2Unidad de Eritropatología. CDB-IDIBAPS. Hospital Clínico y Provincial de Barcelona. España.
Introducción: La hemoglobina inestable “Newcastle” es una enfermedad rara producida por la mutación en el codón 92 del gen de la betaglobina. Hasta el momento se ha descrito en pacientes de origen inglés y ruso. Se manifiesta como anemia hemolítica crónica, agudizada por la sustancias oxidantes (generalmente fármacos) o infecciones.
Caso clínico: Niña de 5 años nacida en Cataluña, sin antecedentes de interés, que consulta por fiebre tratada con ibuprofeno y carbocisteína/prometazina. Exploración física: palidez cutáneo-subictericia; esplenomegalia, no hepatomegalia. Analítica sanguínea: hemoglobina 7,9g/dl, hematocrito 27%, VCM 93,4fl; reticulocitos 30%o. Leucocitos y plaquetas: normales. Anisocitosis, policromasia linfocitos estimulados en sangre periférica; Coombs directo: negativo. AST 69 U/l, total 2,19mg/dl, LDH: 2.489 U/l, haptoglobina < 24,3mg/dl; ferritina 684ng/ml. Serologías negativas. Enzimas eritrocitarias normales. Reinterrogando: la madre está afectada de episodios ictéricos de repetición, con crisis de eritroblastopenia a los 22 años que requirió esplenectomía. Estudio de hemoglobinopatía: test de inestabilidad térmica positivo, sin cuerpos espontáneos. Estudio electroforético y cromatográfico de alta resolución: hemoglobina identificable correspondiente al 12% del total. La secuenciación del gen de la betaglobina heterocigosis de la mutación correspondiente a la hemoglobina “Newcastle”, identificada en la madre; estudio del padre, normal.
Comentarios: Hay que considerar el diagnóstico ante una anemia hemolítica crónica o intermitente con familiar. La colaboración con un laboratorio de eritropatología especializado nos ha permitido identificar una hemoglobinopatía rara, no catalogada anteriormente, y que es la primera descripción de esta hemoglobinopatía en una familia de origen español. La secuenciación genética abre la perspectiva de diagnóstico y posibles futuros tratamientos de este tipo de patologías.
SÍNDROMES HEMOFAGOCÍTICOS. EXPERIENCIA EN 22 CASOSJ.L. Dapena, C. Díaz de Heredia, P. Bastida, A. Llort, I. Elorza y J. Sánchez de Toledo
Servicio de Oncohematología Pediátrica. Hospital Materno-Infantil Vall d'Hebron. Barcelona. España.
Introducción: El síndrome hemofagocítico se caracteriza por una activación y proliferación incontroladas de histiocitos y linfocitos T, con un estado de hipercitoquinemia responsable de los principales signos biológicos. Existen dos formas: la familiar (LFH) y la secundaria. Su diagnóstico puede constituir un verdadero reto para los pediatras.
Objetivo: Valoración clínica y evolutiva de los casos diagnosticados en nuestro centro entre los años 1995 y 2007.
Material y métodos: Revisión retrospectiva, con recogida de datos a través de las historias clínicas, de los 22 pacientes diagnosticados en dicho período según los criterios del HLH-94 y el HLH-2004).
Resultados: 22 pacientes diagnosticados de síndrome hemofagocítico: 5 casos de LFH, 12 casos asociados a infección (IASH) (leishmania, VEB, parvovirus, VHS-6), 2 casos asociados a AR y CU (SAM), 3 casos asociados a enfermedades malignas (MASH). En la LFH la edad en el momento del diagnóstico fue de 2,5 meses (a excepción de un paciente adolescente con la mutación UNC13D). En las formas secundarias el rango osciló entre los 9 meses y los 14 años (mediana de 4 años). Sin diferencias en cuanto al sexo. En el 71% los pacientes eran previamente sanos. El motivo de consulta principal fue la fiebre prolongada (64%). Sintomatología: fiebre (100%), hepatoesplenomegalia (85%), adenopatías (21%), palidez (21%), rash (14%) y alteraciones neurológicas (14%). Laboratorio: citopenia (100%), hipertrigliciridemia (93%), hiperferritinemia (85%), transaminitis (78%), alteración de la coagulación (64%) e hipofibrinogenemia (35%). Ausencia de actividad de las células NK en el 100%. Se observó hemofagocitosis en la médula ósea de la mayoría de los pacientes, y se precisó biopsia hepática o ganglionar en dos. Evolución: recibieron tratamiento según protocolos HLH-94 y HLH-04 9 pacientes: 5 LHF, 2 secundarios a VEB, 1 a herpes virus 6 y 1 a una CU. En 6 de estos 9 casos se realizó posteriormente un trasplante de progenitores hematopoyéticos con evolución favorable en 2 pacientes (dos formas familiares). En el resto de casos se realizó tratamiento etiológico/soporte con un desenlace satisfactorio en 10 pacientes.
Conclusiones: La LFH se diagnostica antes de los 2 años de edad, aunque el importante desarrollo de los estudios genéticos nos permiten detectar formas familiares de inicio más tardío.
Es importante un reconocimiento precoz del problema, lo cual no resulta fácil en ocasiones, para poder instaurar el tratamiento más adecuado a cada situación.
El tratamiento inmunomodulador y el trasplante de progenitores hematopoyéticos constituyen la base del tratamiento de las formas familiares. En nuestra serie, 2/5 LFH sobreviven tras el TPH.
EFECTO INJERTO CONTRA LINFOMA DE HODGKIN TRAS TRASPLANTE ALOGÉNICO DE MÉDULA ÓSEA EN ADOLESCENTES. Brió, I. Badell, M. Torrent, N. Pardo, M. García y J. Cubells
Servicio de Pediatría del Hospital de la Santa Creu i Sant Pau. Universidad Autónoma de Barcelona. España.
Introducción: El linfoma de Hodgkin comprende el 6,6% de los cánceres pediátricos. La supervivencia global de la enfermedad oscila entre el 85 y el 95 % en estadios tempranos, pero empeora en los estadios desfavorables o en las recaídas. Presentamos el caso clínico de un paciente que presentó una segunda recaída tras un trasplante autólogo de médula ósea. En situación de enfermedad residual activa, se realizó un trasplante alogénico de hermano HLA compatible beneficiándose del efecto injerto contra enfermedad.
Caso clínico: Adolescente de 18 años diagnosticado en enero de 2000 (con 11 años) de linfoma de Hodgkin (LH) tipo esclerosis nodular estadio IIIB, a raíz de un cuadro de adenopatías laterocervicales, febrícula y tos seca. Recibió tratamiento quimioterápico y radioterápico según protocolo SEOP EH-2/97: presentó recidiva en adenopatía laterocervical en febrero de 2001. Recibió un trasplante autólogo de progenitores hematopoyéticos de sangre periférica en segunda remisión completa en diciembre de 2001 (acondicionamiento con ciclofosfamida, Vp-16 y BCNU). En septiembre de 2003 se comprueba segunda recidiva con nódulos hepáticos y pulmonares. Se realizan dos tandas de quimioterapia con cisplatino, Vp-16 y Ara-C, con respuesta parcial, y se somete a un trasplante alogénico de progenitores hematopoyéticos de sangre periférica en marzo de 2004 a partir de hermano HLA compatible 9/10 (diferencia en un antígeno HLA A). Se efectúa acondicionamiento de intensidad reducida con fludarabina y melfalán. Infusión de progenitores hematopoyéticos de sangre periférica con un contenido de 8,6 × 10e8 células nucleadas/kg, 18,1 × 10e6 CD34/kg y 7,3 × 10e8 CMN/kg. Presenta quimera total por estudio de ADN desde el día +14 hasta la actualidad, enfermedad injerto contra huésped (EICH) agudo grado II cutáneo y digestivo, que respondió adecuadamente a corticoterapia. En la actualidad, EICH crónico oral y digestivo controlado con budesonida oral. La TC pulmonar a los 3 meses postrasplante muestra desaparición de la enfermedad pulmonar. Persiste en remisión completa a 3 años del trasplante.
Conclusiones: El trasplante alogénico de médula ósea se asocia a un efecto clínico antineoplasia independiente de la acción del acondicionamiento pretrasplante por mecanismos hasta ahora desconocidos. Se ha descrito ampliamente el efecto del injerto contra enfermedad en leucemias aguda y en leucemias crónicas, pero no está tan descrito el efecto injerto contra tumor en los linfomas.
COMPLICACIONES NEUROLÓGICAS ASOCIADAS AL TRATAMIENTO DE LA LEUCEMIA LINFOBLÁSTICA AGUDA: EXPERIENCIA DE UN CENTROM. López Duarte1, E. Bureo1, L. Yáñez1, A. Uresandi1, E.M. Rodríguez Rodríguez2, M. Puente2, B. González-Mesones2 y A. Iriondo1
1Servicio de Hematología-Hemoterapia. 2Servicio de Neurología. Hospital Universitario Marqués de Valdecilla. Santander. España.
Introducción: En nuestro centro hemos diagnosticado en los últimos 10 años 33 leucemias linfoblásticas agudas (LLA), de las cuales 8 han sido en pacientes entre 10–16 años. Todos los pacientes se han incluido en los sucesivos protocolos SHOP 94/99/05. Describimos 2 casos de complicaciones neurológicas objetivadas entre estos pacientes y presuntamente atribuibles al tratamiento con altas dosis de metotrexato (MTX) y L-ASA, respectivamente.
Descripción de los casos: El primer caso es una paciente mujer de 15 años diagnosticada en julio de 2005 de LLA B-II (EGIL) My+ y con expresión aberrante de CD2+, con reordenamiento de IgH positivo (Southern blot), alcanzando remisión completa morfológica y por inmunofenotipo tras la inducción según protocolo SHOP 2005. Posteriormente, recibió los primeros 2 ciclos de consolidación con MTX en altas dosis y triple terapia intratecal (TIT) presentando toxicidad de grado 2 de la OMS en el hígado tras el primero. A los 14 días de la TIT y 16 tras esta dosis de MTX ingresa por cuadro consistente inicialmente en debilidad en miembro superior izquierdo (MSI) con progresivo desarrollo de tetraparesia ESI: 2/5, EII: 0/5, EID: 4/5, ESD: 4/5) y hemihipoestesia izquierda (facio-braquio-crural), disartria y desviación de la comisura bucal. En la exploración física: paresia facial central derecha, disartria grave, reflejo cutáneo plantar extensor izquierdo. En la RM se observan dos lesiones en espejo, parietales hipointensas en T1, hiperintensas en secuencias T2 y FLAIR sin captación de contraste con defecto de difusión, compatibles con leucoencefalopatía multifocal tóxica. Tras tratamiento con ácido folínico (15mg/día) y terapia conservadora se objetiva mejoría lentamente progresiva en 5–7 días. En la actualidad está en la fase de mantenimiento del segundo año, en remisión completa, presentando desde el punto de vista neurológico hemiparesia izquierda de predominio distal (4/5) residual con temblor fino en MSI. A partir del efecto adverso descrito se suprimió del tratamiento tanto el metotrexato intratecal como sistémico. El segundo caso es un paciente varón de 12 años diagnosticado en marzo de 2006 de LLA-BII sin alteraciones en el estudio biológico, de muy alto riesgo por persistencia de alta tasa de enfermedad residual (2,6% blásticas por inmunofenotipo en el día +28 y > 0,1% en la consolidación). En el día +21 (tras la segunda dosis de L-ASA), el paciente acude por episodio de crisis convulsivas tónico-generalizadas con hemiplejia derecha acompañada de afasia motora. En la TC craneal se objetivan pequeñas hemorragias bifrontales, la mayor de 36mm de diámetro. En la RM realizada posteriormente se comprueba la existencia de hematomas de localización bifrontal e imágenes sugestivas de trombosis del seno sagital superior. Analíticamente, se objetivó alargamiento del TTPA e hipofibrinogenemia marcada (53mg/dl). Es homocigoto C/C en la posición 1298 para el gen de la MTHFR. El cuadro clínico presentó mejoría progresiva (EID: 4/5 y disfasia motora leve-moderada) tras 10 días de anticoagulación con heparina sódica intravenosa, resolviéndose casi en su totalidad tras los primeros 30 días. En la actualidad se encuentra en el tercer mes de un trasplante alogénico de donante no emparentado, acondicionado con irradiación corporal total fraccionada y ciclofosfamida, en remisión completa y sin déficit neurológico asociado.
Conclusión: El metotrexato es un fármaco eficaz en el tratamiento de la LLA, con notables efectos tóxicos en el SNC, y la leucoencefalopatía es su forma más frecuente. La necesidad de modificar el tratamiento previsto no está claramente establecida. También se desconoce la dosis de leucovorín necesaria para revertir los efectos tóxicos del fármaco. La mayor parte de los cambios observados en la RM se resuelven con el tiempo. Por otro lado, el riesgo de trombosis en la LLA es del 1-37% (el 5% según un estudio reciente). La asociación de L-ASA de E. coli, prednisona y factores protrombóticos previos dispara este riesgo. El desarrollo y la gravedad de estas complicaciones neurológicas asociadas al tratamiento de la LLA nos plantea la necesidad de establecer estrategias preventivas antitrombóticas. Queda por determinar el efecto de estos cambios en el desarrollo cognitivo y en la calidad de vida de estos pacientes.
TRASPLANTE DE PROGENITORES HEMOPOYÉTICOS EN NIÑOS CON SÍNDROME DE HURLERJ. Dapena, C. Díaz de Heredia, D. Vizmanos, T. Olivé y J. Sánchez de Toledo
Servicio de Oncología-Hematología Pediátrica. Hospital Vall d'Hebron, Bacelona. España.
Introducción: El síndrome de Hurler es un error innato del metabolismo. El déficit de la enzima α-L-hialuronidasa produce una acumulación de heparina y dermatán sulfatos, causantes de la hepatoesplenomegalia, las anomalías esqueléticas, la miocardiopatía y el deterioro neurológico progresivo, que lleva generalmente a una muerte temprana antes de los 8 años. El trasplante de progenitores hemopoyéticos (TPH) puede mejorar el curso clínico.
Material y métodos: Presentamos a los 8 pacientes con síndrome de Hurler (4 niños y 4 niñas) sometidos a TPH en nuestro centro. La edad media al diagnóstico fue de 10 meses (6 meses-5 años). De los 8 pacientes, 6 presentaban fenotipo grave y 2, moderado. Tres pacientes recibieron terapia sustitutiva con la enzima, previa al TPH. La mediana de la edad en el momento del trasplante fue de 26 meses (17 meses-10 años). Cuatro pacientes fueron sometidos a TPH de donante hermano HLA-idéntico, y 4 a TPH de donante no emparentado (DNE) (2 de médula ósea (MO) y 2 de sangre de cordón umbilical [SCU]). El régimen de acondicionamiento consistió en BU 16–20mg/kg y CY 200mg/kg en los TPH de hermano HLA-idéntico, y BU + CY + ATG o irradiación nodal total en los TPH de DNE. La profilaxis del EICH consistió en CyA + MTX en los TMO y en CyA + prednisona en el TSCU. Todos los pacientes injertaron excepto uno, que falleció por progresión de la enfermedad; 5 pacientes presentaron EICH de grado I y 2 de grado II. Ningún paciente desarrolló EICH crónico; 7 de los pacientes están vivos con quimerismo completo del donante, y niveles de α-L-hialuronidasa normales o comparables con las concentraciones del donante. Las visceromegalias que se observaban en el momento del trasplante en 4 pacientes remitieron. La miocardiopatía presente en 5 de los pacientes mejoró o se estabilizó. Las anomalías esqueléticas no mejoraron en ninguno de los pacientes. El desarrollo neurológico de los pacientes sometidos a trasplante antes de los 2 años fue normal. En los pacientes trasplantados entre los 2 y los 3 años de edad, el daño neurológico no progresó. Finalmente, 2 pacientes con fenotipo moderado, trasplantados a los 5 y 10 años de edad, respectivamente, no desarrollaron posteriormente un daño neurológico grave.
Conclusión: Los pacientes afectados de síndrome de Hurler tienen una supervivencia prolongada si son sometidos a TPH, y si éste se realiza de forma temprana, no presentarán posteriormente deterioro neurológico. La SCU ha demostrado ser una fuente válida de progenitores hemopoyéticos para el trasplante en estos pacientes.
REMISIÓN PROLONGADA CON CLADRIBINA EN HISTIOCITOSIS DE CÉLULAS DE LANGERHANS REFRACTARIA AL TRATAMIENTO CONVENCIONALM. Torrent, I. Badell, N. Pardo, M. García, S. Brió, B. Pineda, E. López, R. Álvarez, E. Turón y G. Fraga
Servicio de Pediatría. Hospital de Sant Pau. Barcelona. España.
Objetivo: Los pacientes afectados de histiocitosis de células de Langerhans (HCL) que se muestran refractarios al tratamiento inicial o que recidivan precozmente tienen un mal pronóstico. En los últimos años se han ensayado distintos tratamientos de rescate alternativos para estos pacientes, entre ellos la cladribina. La cladribina o 2-cloro-deoxiadenosina (2-CdA) es un análogo de las purinas que interfiere en la reparación del ADN y acelera el proceso de apoptosis.
Material y métodos: Paciente de 17 meses de edad que consulta por tumoración en el paladar y lesiones eczematosas en el abdomen y la zona del pañal. En el aspecto analítico destaca anemia inflamatoria. Se efectúa RM macizo-facial que muestra masa en el seno maxilar derecho que invade celdillas etmoidales y el suelo orbitario, con estructuras óseas del maxilar erosionadas y compromiso del músculo recto inferior; hay presencia de adenopatías bilaterales. Se efectúa biopsia de la masa + PAAF de adenopatías y punch de lesiones cutáneas. La anatomía patológica es compatible con histiocitosis de células de Langerhans (HCL): proliferación de células de Langerhans con intensa expresión citoplasmática de CD1a, CD68 y S-100. Estudio de extensión negativo. Se inicia tratamiento con vinblastina, etopósido, mercaptopurina y prednisona según protocolo nacional SEOP-HCL 2000 con desaparición de las lesiones cutáneas tras el primer ciclo de tratamiento y respuesta parcial inicial de la tumoración maxilar. A los 2 años y 2 meses se objetiva en una TC de control progresión tumoral con importante afectación de ambos senos maxilares. Se inicia tratamiento de rescate con cladribina a 5mg/m2/día durante 5 días por ciclo y por un total de 6 ciclos. Buena respuesta: desaparición total de la infiltración y excelente tolerancia al tratamiento. Último ciclo de cladribina en enero de 2005.
Resultados: Se ha mantenido desde el final del tratamiento asintomática y sin evidencia de enfermedad por estudios de imagen repetidos (TC y PET). Presenta como secuela pérdida parcial de la dentición.
Conclusiones: La 2-CdA ha mostrado su eficacia en el tratamiento de procesos linfoproliferativos de curso crónico en adultos y en la tricoleucemia. También se ha ensayado en pacientes afectados de HCL de mala evolución por su capacidad de producir monocitopenia y se han descrito remisiones duraderas en el 50-70% de los casos.
SÍNDROME DE NOONAN Y ALTERACIONES HEMATOLÓGICASM. Forns, A. Morales, A. Llort, P. Bastida y J. Sánchez de Toledo
Servicio de Oncología-Hematología Pediátrica. Hospital Vall d'Hebron. Barcelona. España.
Objetivos: Descripción de 2 casos pediátricos afectados de síndrome de Noonan que presentan alteraciones hematológicas características de dicho síndrome.
Resultados:1) Varón de 3 meses de edad ingresa para estudio de hepatoesplenomegalia, mielomonocitosis y plaquetopenia. Antecedentes: estrés respiratorio neonatal, orientado como síndrome de aspiración meconial. Fenotipo con turricefalia, hipertelorismo, epicanto, orejas de baja implantación, cuello alado, mamilas muy separadas, pectus carinatum, criptorquidia bilateral y sindactilia del tercer dedo del pie. Cardiopatía congénita: estenosis pulmonar y comunicación interauricular. Bronquiolitis a los 2 meses de vida detectándose alteraciones hematológicas.
Exploración física a su ingreso: fenotipo descrito anteriormente, palidez mucocutánea, soplo eyectivo IV/VI de predominio en foco pulmonar, hepatomegalia (5cm), esplenomegalia dura (10cm). Exploraciones complementarias: hemograma: hematocrito: 30,8%; hemoglobina: 10g/dl; leucocitos: 35,9 × 109/l (neutrófilos 18%; linfocitos 52%; monocitos 20%); plaquetas 94 × 109/l.
Morfología de sangre periférica (sp): precursores hematopoyéticos mieloides (2%), eritroblastosis (6%). Médula ósea (MO): tres series hematopoyéticas en todos los estadios madurativos, blastos (5 %) morfología variada, monocitosis (5 %), linfocitosis (29%). PCR CMV: 120.000 copias.
Diagnóstico: síndrome de Noonan, infección por CMV y leucemia mielomonocítica juvenil (LMMJ) frente a síndrome mieloproliferativo transitorio. Recibe tratamiento con foscarnet intravenoso.
Tras la erradicación de CMV en sangre se observa recuperación del número de plaquetas, así como disminución en la cifra de leucocitos, aunque persiste monocitosis marcada. Disminuye también la esplenomegalia en tamaño y consistencia. Actualmente sigue controles ambulatorios (1,5 meses del diagnóstico). 2) Varón de 22 meses ingresa para estudio de leucocitosis. Antecedentes: sospecha de sepsis neonatal tardía no confirmada en la que se detecta alteración hematológica. Exploración física: fenotipo peculiar con orejas de implantación baja y eje visual antimongoloide; el resto de la exploración es normal. Exploraciones complementarias: hemograma: hematocrito: 36 %; hemoglobina: 13g/dl; leucocitos: 37,7 × 109/l (neutrófilos 31%; linfocitos 41%; monocitos 13%); plaquetas 200 × 109/l). Citometría de flujo en SP: monocitos maduros (16%), promonocitos (7%), blastos mieloides (6,5%). Médula ósea (MO): celularidad medular conservada con escasos precursores de las tres series, destaca infiltración por células immaduras con dos poblaciones, linfocitosis (29%), monocitosis (15%). Biología molecular en MO: PCR-RT reordenamiento BCRABL ausente. Determinación PTPN11: positiva. PCR virus: negativa. Ecocardiograma: normal. Diagnóstico: síndrome de Noonan y LMMJ frente a síndrome mieloproliferativo transitorio. Ante el buen estado clínico y la ausencia de cambios analíticos, actualmente sigue controles ambulatorios (1,5 meses del diagnóstico). El cultivo in vitro de progenitores mieloides de SP en ambos pacientes ha demostrado un patrón de crecimiento compatible con LMMJ que se debe valorar en el contexto clínico del paciente.
Conclusiones: Estos 2 casos clínicos muestran la asociación ya descrita entre el síndrome de Noonan y hemopatías malignas, muestran además la dificultad en el diagnóstico diferencial entre LMMJ y SMP transitorio. El primer caso también muestra la similitud en la forma de presentación entre dichas hemopatías y ciertas infecciones microbianas diseminadas.
RESPUESTA A IMATINIB E INFUSIÓN DE LINFOCITOS DEL DONANTE EN UNA LLA PHILADELPHIA EN RECIDIVA PRECOZ: A PROPÓSITO DE UN CASOM. López Duarte, E. Bureo, M.A. Cuadrado, M. Albajar, A. Uresandi, L. Yáñez, M. Puente, B. González-Mesones y A. Iriondo
Servicio de Hematología-Hemoterapia. Hospital Universitario Marqués de Valdecilla. Santander. España.
Introducción: La incidencia de leucemia linfoblástica aguda Philadelphia (LLA Ph) en la edad pediátrica es del 3–5 % de los casos. Hasta el momento actual, el trasplante alogénico suponía la única opción curativa de estos pacientes. La tasa de recidivas postrasplante alogénico es de hasta el 20%, describiéndose ratios inferiores (3–5 %) en la era del imatinib. El tratamiento de las recidivas postrasplante no está establecido. Describimos nuestra experiencia reciente en el tratamiento de esta situación clínica.
Descripción del caso: Niño de 8 años diagnosticado en marzo de 2005 de leucemia linfoblástica aguda pre-B (BII) Philadelphia con reordenamiento bcr-abl positivo en las isoformas p190 y p210. Se realizó inicialmente tratamiento poliquimioterápico según protocolo SHOP-MAR-99 con persistencia de enfermedad mínima residual (EMR) > 1 % por inmunofenotipo tras el ciclo de inducción.
Por este motivo, posteriormente se asoció imatinib en dosis de 260mg/m2 al tratamiento poliquimioterápico según el protocolo Pethema LAL-Ph. Tras completar el primer bloque de consolidación y el primer ciclo del segundo bloque asociados al imatinib, se consigue un buen control de la enfermedad con < 0,1 % de EMR por citometría de flujo, negativización de la p190 por RT-PCR pero persistencia de la p210. En septiembre de 2005 recibió un trasplante alogénico de células progenitoras hematopoyéticas (TCPH) de médula ósea de donante no emparentado, acondicionado con ciclofosfamida y radioterapia corporal total fraccionada. En el día +60 se confirma remisión completa (RC) molecular y se inicia descenso progresivo de la ciclosporina a partir del día +80 ante la ausencia de enfermedad injerto contra huésped (EICH).
En febrero de 2006 (5o mes pos-TCPH) se objetiva recidiva testicular y se observa en la médula ósea el 0,8% de población leucémica por inmunofenotipo (IF) y con RT-PCR positiva para p210 (p190 negativa), a pesar de quimerismo linfohematopoyético 100% del donante. En esta situación, se procedió a la retirada brusca de la ciclosporina, se inició de nuevo imatinib en las dosis señaladas con ascenso posterior en función de la tolerancia hematológica hasta una dosis máxima de 400mg/día y se realizó además orquiectomía izquierda y RT locorregional y posteriormente ante la ausencia de EICH, infusión de linfocitos del donante (ILD) en dosis crecientes (2,5 × 10E7/kg, 1 × 10E8/kg, 1,5 × 10E8/kg). Para prevenir la recidiva en el líquido cefaloraquídeo (LCR), se administraron 4 intratecales con dosis menores de metrotexato y las dos últimas sin éste para disminuir el riesgo de leucoencefalopatía multifocal progresiva secundaria. Tras el primer mes persiste aún EMR por IF (0,12%), consiguiéndose RC molecular entendida por la ausencia de detección en la RT-PCR cualititativa al cuarto mes. Actualmente, se encuentra en el mes 30 post-TCPH (14 meses tras la recidiva) en remisión completa molecular y está incorporado a su actividad habitual, escolar y deportiva.
Comentarios: El Imatinib ha demostrado un papel relevante en el control de la LLA Philadelphia de reciente diagnóstico o en recidiva, aunque generalmente de efecto transitorio. La combinación de radioterapia local, retirada de la inmunosupresión, profilaxis del SNC, imatinib e infusión de linfocitos del donante ha conseguido hasta ahora un control excelente de la enfermedad en nuestro paciente, evitando la utilización de quimioterapia sistémica en el contexto de recidiva precoz pos-TCPH. La coexpresión de mARN de p190 y p210 ocurre hasta en el 50-70% de las leucemias mieloides crónicas, aunque menos frecuente y estudiado, está descrito en las LLA, lo que obliga a su detección simultánea en el momento del diagnóstico y de la recidiva. Las implicaciones biológicas de este fenómeno aún están por determinar.
LEUCEMIA MIELOIDE CRÓNICA: LA EXPERIENCIA EN NUESTRO CENTROA. Moreno Álvarez, L. Fernández Pérez, A. Prado Carro, A. Ávila Álvarez, S. Arnaiz Pena, I y Rodríguez Sánchez
Servicio de Pediatría. Complejo Hospitalario Juan Canalejo. A Coruña. España.
Introducción: La leucemia mieloide crónica (LMC) es un trastorno clonal del tejido hematopoyético que representa el 2–3 % de los casos de las leucemias en la infancia. Está asociada a una traslocación específica, t(9:22) (q34;q11), que se denomina cromosoma Filadelfia. Se desarrolla en tres fases clínicas: crónica, acelerada y crisis blástica.
Objetivos: Describir las características de los casos presentados en nuestra unidad y analizar la respuesta al tratamiento con el inhibidor de transducción de señal de la proteína tirosincinasa bcr-abl.
Material y métodos: Revisión retrospectiva de los casos diagnosticados entre los años 1999 y 2006.
Resultados: Caso 1. Niña de 10 años asintomática remitida para estudio por su pediatra por presentar en analítica de sangre displasia de la serie blanca. Exploración: dentro de la normalidad salvo eritema con micropápulas en región facial y en miembros inferiores presente desde el período de lactante. Analítica: hemoglobina 12,4g/dl; plaquetas: 440.000; leucocitos: 35.400 (mielocitos 3 %, metamielocitos 3 %, cayados 4 %, segmentados 68 %, basófilos 3%, linfocitos 14%, monocitos 5%). Morfología: serie roja anisocitosis; serie blanca leucocitosis con mielemia y basofilia; displasia de plaquetas. LDH y vitamina B12 normales, FAG: 24.
Caso 2. Niño de 13 años con anorexia de 2 meses de evolución remitido igualmente por su pediatra para estudio. Exploración: piel cetrina con palidez discreta de mucosas, microadenopatías laterocervicales, axilares e inguinales y esplenomegalia. Analítica: hemoglobina 8g/dl; hematocritos 24 %; plaquetas 948.000; leucocitos: 325.000 (neutrófilos 86%, linfocitos 3%, monocitos 0,6%, basófilos 10%). Morfología: mielemia con basofilia; serie granulocítica mal segmentada y degranulada; serie roja policromasia y punteado basófilo. LDH 1.500, vitamina B12 > 200, F.A.G.: indetectable. Evolución: la biopsia de médula ósea mostró en ambos casos hiperplasia granulocítica y megacariocítica con basofilia y eosinofilia moderada. El estudio de genética molecular (FISH) evidenció el reordenamiento bcr-abl. Ninguno de los pacientes tenía donante HLA compatible.
Tratamiento: Ambos pacientes reciben tratamiento con imatinib y se encuentran actualmente asintomáticos. El primer caso se trató previamente con interferón alfa e hidroxiurea. Tras dicho tratamiento, el estudio de genética molecular no muestra el reordenamiento bcr-abl.
Conclusiones: La mala regulación de la actividad cinasa bcr-abl es la principal responsable de la transformación de las células hematopoyéticas en LMC. Este descubrimiento sienta las bases para buscar dianas terapéuticas dirigidas específicamente contra las alteraciones genéticas responsables de la transformación clonal; como es el caso del Imatinib. En nuestros pacientes la tolerancia al tratamiento fue adecuada, tanto desde el punto de vista clínico (fármaco bien tolerado y sin presentar efectos secundarios), analítico y de la genética molecular. Sin embargo, aspectos por definir son la aparición de resistencias, la asociación con otros fármacos y la duración necesaria del tratamiento para la remisión citogenética y molecular definitiva.
CÁNCER EN EL ADOLESCENTE: EXPERIENCIA DE UN CENTROF. Lendínez Molinos, M.A. Vázquez López, M. Leyva Carmona, M. Rodríguez, P. Aguilera, P. Oliva y A. Bonillo
Unidad de Oncología Pediatrica. Servicio de Pediatría. Hospital Torrecárdenas. Almería. España.
Introducción: El diagnóstico de cáncer en el adolescente exige un abordaje distinto que en la edad infantil. La aceptación del tratamiento oncológico, sus complicaciones y secuelas, el cambio de la imagen corporal y el futuro incierto derivado del pronóstico de una enfermedad grave son realidades a las que tiene que hacer frente el adolescente y que hace imprescindible la consideración de equipos multidisciplinares específicos.
Objetivo: Describir las características de los casos de cáncer en adolescentes diagnosticados en nuestro servicio y analizar la evolución y supervivencia.
Material y métodos: Se revisan las historias clínicas de los pacientes diagnosticados de cáncer entre enero de 1980 y junio de 2006 que precisaron tratamiento oncológico en la adolescencia (13–18 años). Se incluyeron 19 nuevos diagnósticos, 2 casos de recidivas tardías y 5 segundas neoplasias. Analizamos las características clínicas, el tipo de neoplasia, la evolución y la supervivencia global utilizando la curva de Kaplan-Meier.
Resultados: La edad media del total de los 26 pacientes incluidos fue de 13,9 ± 12,8 años. Fueron varones 18 (69%). La distribución según el tipo de neoplasia fue: leucemias 7 casos (27%), óseos 6 casos (23%), SNC 4 casos (15.4%), linfomas 3 casos (11,5%), neuroblastomas y sarcomas 2 casos respectivamente (7,7%), un carcinoma de tiroides (3,4%) y un melanoma (3,4%). Las recidivas tardías correspondieron a un neuroblastoma y una LLA. Los segundos tumores correspondieron a LMA tras LNH, 1 sarcoma tras meduloblastoma, un tumor óseo tras LLA y 2 meningiomas tras LLA. El tiempo de latencia medio fue de 8,9 años (de 0,5 a 15 años). Han recidivado 5 casos (19,2%) y han fallecido 10 casos (38,5%), 4 por causa infecciosa (40%) y 6 por enfermedad progresiva (60%). Un total de 8 casos presentan algún tipo de secuela. Precisaron asistencia psicológica 10 casos. La supervivencia global tras 5 años de seguimiento es del 52,5%.
Conclusiones: Las leucemias y tumores óseos fueron los más frecuentemente observados. Las secuelas se presentan en una proporción elevada. El diagnóstico de cáncer en el adolescente debe considerar el apoyo psicológico en todos los casos, sobre todo en aquellos pacientes con experiencia oncológica previa (recidivas y segundas neoplasias). La supervivencia obtenida fue inferior a la descrita para menores de 15 años. Mejorar el pronóstico del cáncer en el adolescente debe ser la meta de todo equipo multidisciplinar.
LEUCEMIA MIELOIDE CRÓNICA EN LA ADOLESCENCIA: ¿IMATINIB COMO PRIMERA ALTERNATIVA?S. Brió, I. Badell, N. Pardo, M. Torrent, M. García, P. Nomdedeu, R. Álvarez, E. Turon y J. Cubells
Servicio de Pediatría del Hospital de la Santa Creu i Sant Pau. Universitat Autónoma de Barcelona.
Introducción: La leucemia mieloide crónica es una enfermedad poco frecuente en la adolescencia. Imatinib es una proteína tirosin-quinasa inhibidora selectiva del gen BCR-ABL con actividad frente c-abl, bcr/abl, c-kit y sobre el receptor del factor de crecimiento plaquetar. Está indicado en el tratamiento de cualquier fase de la leucemia mieloide crónica, en fase blástica, aguda o crónica. Presentamos el caso de un adolescente en tratamiento con Imatinib como primera elección tras el diagnóstico.
Caso clínico: Paciente de sexo masculino de 13 años de edad, sin antecedentes patológicos de interés ni sintomatología previa, a quien por una lesión cutánea se le practica hemograma que muestra leucocitos: 109,5 × 10E9/l con predominio de elementos mieloides no blásticos del orden de 6% promielocitos, 22% mielocitos, 9 % metamielocitos, serie roja normal y recuento plaquetar: 522 × 10E9/l. Estudio de coagulación: normal y bioquímica: LDH: 858 U/l, resto normal. En la exploración física no se observan adenopatías ni organomegalias. Una ecografía abdominal informa de tamaño de bazo en límites superior de normalidad.
El aspirado medular demuestra marcado aumento de la celularidad, hiperplasia mieloide, sin hiato ni exceso de blastos. Se informa de leucemia mieloide crónica en fase crónica. Cariotipo: 46XY, t(9,22)(q34;q11,2). Estudio molecular del reordenamiento BCR/ABL (región génica P210) correspondiente a una traslocación t(9;22) del tipo b2a2 (cromosoma philadelphia) resulta positivo. Se inicia tratamiento con hidroxiurea, hidratación y alopurinol oral con respuesta parcial. En septiembre de 2004 se inicia tratamiento con Imatinib (400mg/día) y se realiza monitorización trimestral con PCR cuantitativa en médula ósea/sangre periférica de BCR/ABL para valoración de la respuesta, manteniéndose en remisión.
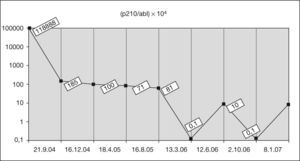
Conclusiones: Desde la aparición de Imatinib, el trasplante alogénico de médula ósea ha quedado en segundo plano en el tratamiento de la leucemia mieloide crónica. Nuestro paciente se mantiene en remisión molecular a 2 años y 6 meses de inicio del tratamiento.
