















Pósters
El análisis de la secuencia del genGH1demuestra una elevada variabilidad
C. Quinteiro García 1, L. Castro-Feijoó 2, L. Loidi 1, J. Barreiro 2, C. Redondo 1, D. González-Lestón 3, R. Cañete Estrada 4 y P. Fariña 1
1Fundación Pública Galega de Medicina. 2Fundación Pública Galega de Medicina Xenómica. 3Unidad de Endocrinología. Santiago de Compostela. España.
Introducción: La mayoría de los casos de talla baja asociada a deficiencia de GH ocurren de forma esporádica e idiopática, pero entre el 5 y el 30 % de los pacientes tienen un familiar en primer grado con el mismo fenotipo, que nos indica una posible etiología genética. La confirmación de esta base genética viene dada por la demostración de mutaciones heredables en el gen GH1 en individuos con retraso en el crecimiento. Hasta ahora el estudio molecular del gen GH1 se ha realizado principalmente en pacientes con una deficiencia grave de GH, y aunque se han descrito mutaciones concretas en el gen GH1, en la mayoría de los casos se han encontrado grandes deleciones. Es probable que si se estudiaran molecularmente pacientes con una sintomatología menos grave se podrían encontrar lesiones menos dramáticas que sean la causa del fallo en el crecimiento y que de otra forma habrían quedado sin identificar.
Pacientes y métodos: Se estudiaron 176 pacientes con las siguientes hipótesis diagnósticas: déficit de GH (DGH): 109 pacientes; talla baja familiar (TBF): 38 pacientes, y 29 con otros diagnósticos relacionados con retraso del crecimiento. Las hipótesis diagnósticas fueron proporcionadas por sus propios endocrinólogos pediatras. El análisis del gen GH1 se realizó mediante secuenciación cíclica directa de un fragmento de PCR que engloba toda la secuencia del gen GH1 y regiones colindantes, y búsqueda de grandes deleciones mediante Southern blot y PCR específica de corte enzimático.
Resultados y conclusiones: No se encontraron deleciones en ninguno de nuestros pacientes. En el estudio de la secuencia identificamos 31 cambios nucleotídicos diferentes, más frecuentemente en la región 59 y en los intrones. De éstos, 20 no han sido descritos antes. En la secuencia codificante encontramos 9 mutaciones, 5 sinónimas (Ser111, Ser121, Tyr129, Thr149 y Val199) y 4 de sentido equivocado (Phe118Leu, Ile205Met, Thr3Ala, Thr3Arg y Val136Ile), todas ellas en heterocigosis. Ser111, Phe118Leu, Ser121 y Tyr129 aparecieron juntas en un paciente con diagnóstico de TBF; la Ile205Met se encontró en un paciente con DGH; Thr3Ala en 2 casos de TBF y 6 casos de DGH; Thr3Arg en un caso de TBF; Val136Ile en 2 pacientes con DGH y Val199 en un caso de DGH. Se identificaron un total de 12 cambios diferentes en la región 59 del gen; la mayoría de éstos ocurren en las mismas posiciones en las cuales el gen GH1 difiere de sus genes parálogos, lo que sugiere que pueden originarse por mecanismos de conversión génica. En la secuencia de los intrones identificamos 10 cambios en la secuencia, todos ellos en heterocigosis, excepto la variante P1 del intrón IV (A/T) (rs2665802), que apareció en homocigosis y en heterocigosis en todos los grupos; se ha comunicado que este cambio está asociado con la concentración de secreción de GH y con la altura de los individuos. Encontramos que el 74,3 % de nuestros pacientes con DGH eran portadores de al menos un alelo A, mientras que eran portadores de este alelo el 65,7 % de los pacientes con TBF. Esta diferencia no mostró significación estadística quizá por el bajo número de pacientes estudiados con TBF. Con este estudio hemos encontrado que el gen GH1 presenta muchas variantes con un espectro continuo de sus cambios nucleotídicos desde polimorfismos aparentemente neutrales a variantes disfuncionales a lo largo de todo el gen con dos zonas calientes una en la región 59 y la otra en el intrón IV. Algunas de estas variantes parecen ser originadas por un mecanismo de conversión, consecuencia en sí misma de la estructura multigénica del locus GH.
Activación neuronal hipotalámica en la respuesta somatotropa a estímulos combinados de GHRH y secretagogos
L. Jiménez Reina, M.D. Cañete Vázquez, E. García Martínez, J.P. Rojas Ramírez, G. Bernal Valls y R. Cañete Estrada
Endocrinología Pediátrica y Departamento. Facultad Medicina y Hospital Universitario Reina Sofía. Córdoba. España.
Introducción: En el contexto de estudio de reserva hipofisaria en ratas prepúberes se evalúan las implicaciones hipotalámicas de distintos núcleos en el control de la secreción de hormona de crecimiento (GH).
El propósito de este trabajo es analizar, en animales prepúberes, la respuesta de las neuronas hipotalámicas de los núcleos arcuato y paraventricular tras la administración de diferentes estímulos de GH: somatostatina (SRIH), hormona estimuladora de GH (GHRH), GHRP-6, ghrelina y tratamientos combinados entre ellos.
Material y métodos: Se distribuyeron en 12 grupos ratas Wistar hembras de 30-35 días de vida. De ellas, 6 recibieron SRIH y 6 suero salino a los 90 min. A todos, respectivamente, se les administró, en el tiempo 0 min, suero salino, GHRH, GHRP-6, ghrelina, GHRH + GHRP-6, GHRH + ghrelina (1 μg/kg peso en todos los casos). Los animales se sacrificaron por decapitación a los 90, 0, 30 y 90 min, para obtener los cerebros, que se fijaron en paraformaldehído al 6 % en TBS, con sucrosa al 15 %. Estos se sumergieron en Tissue Tek-OCT®, para su congelación en isopentano (Merck) enfriado en nitrógeno líquido y tras su posterior corte en criostato, fueron inmunoteñidos con antiproteína c-fos, determinándose la proporción de neuronas hipotalámicas de los principales núcleos hipotalámicos.
Resultados: En el núcleo arcuato la activación neuronal se produce con los distintos estímulos de GHRH, GHRP-6, ghrelina, GHRH + GHRP-6 y GHRH + ghrelina y alcanza los mayores valores con los estímulos combinados de GHRH a los 90 min (p < 0,05; 90 min frente a 0 min). El tratamiento previo con SRIH (90 min) induce activación neuronal (p < 0,05; 0 min frente a 90 min); en estas circunstancias, sólo la administración posterior de GHRH + GHRP-6 potencia dicha activación. En el núcleo paraventricular la activación neuronal es más importante en los grupos tratados con ghrelina y su combinación con GHRH, desde los 30 min del estímulo (p < 0,05; 30 y 90 min frente a 0 min). Observamos además una disminución de la expresión de proteína c-fos, con la administración previa de SRIH (p < 0,05; 90 min frente a 0 min).
Conclusiones: En el núcleo arcuato, implicado en el control de GH, se observan modificaciones claras de la activación neuronal tras la administración de los distintos estímulos liberadores de GH. En el núcleo paraventricular poco implicado en el control de la secreción de GH, la respuesta activadora se observa sólo con aquellos estímulos implicados en otras funciones biológicas.
El tratamiento con hormona de crecimiento restaura la expresión de IGF-I en el hipotálamo de rata tras nefrectomía
I. Molinos Norniella, J.A. Chowen, E. Carbajo-Pérez, F. Santos, J. Argente y L.M. Frago
Pediatría. Universidad Autónoma de Oviedo. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Introducción: Estudios experimentales indican que la capacidad de las células pituitarias para secretar hormona de crecimiento (GH) en respuesta al estímulo con hormona liberadora de GH (GHRH) está deprimida en el fallo renal crónico (FRC). Esta disminución se ha demostrado que no es debida a una disminución del número de células somatotropas ni del ARNm del receptor de GHRH en la hipófisis, por lo que podría obedecer a una desregulación inducida por la uremia de los mecanismos hipotalámicos de control.
Objetivos: Conocer la influencia del FRC sobre la expresión hipotalámica de los ARNm de IGF-I y neuropéptido Y (NPY) en un modelo de uremia experimental.
Material y métodos: Se estudiaron los hipotálamos de 4 grupos (n = 19) de ratas jóvenes: FRC, función renal normal con alimentación ad limitum (SAL), función renal normal con alimentación pareada con el grupo FRC (SPF) y FRC tratadas con 10 U/kg/día de GH humana recombinante (FRC + GH) durante una semana. Se extrajo el ARN con el método del Trizol y se hicieron los estudios de RT-PCR en tiempo real con un equipo ABI PRISM 7000. La cuantificación del ARNm se hizo por el método de ΔΔCt (Ct del gen buscado Ct de un gen calibrador). Los primeros usados fueron: IGF-I AS → TCC TTC TGA GTC TTG GGC ATG T; IGF-1 S → CCT ACA AAG TCA GCT CGT TCC A; NPY AS → CAA CGA CAA CAA GGG AAA TGG Y NPY S → CCT GTC CCA CCC AAT GCA.
Resultados: Los animales FRC y FRC + GH tuvieron concentraciones séricas de urea similares y superiores a los grupos SAL y SPF. Los animales FRC fueron hipocrecidos mejorando el retraso de crecimiento en el grupo FRC + GH. Respecto a los niveles de ARNm de IGF-I y de NPY, estableciendo comparaciones entre grupos de los valores CT, se obtuvieron los siguientes resultados:
Para el IGF-1:

Para el NPY:

Conclusiones: El FRC inhibe la expresión del ARNm de IGF-I hipotalámico. Esta inhibición no atribuible exclusivamente a la malnutrición asociada y se normaliza con el tratamiento con GH. Las concentraciones de ARNm de NPY no se modifican significativamente en el FRC.
Concentraciones circulantes de adipocitocinas en la infancia y adolescencia: influencia del desarrollo puberal y sexo
G. Martos Moreno, V. Barrios, L. Soriano-Guillén, J. Pozo y J. Argente
Servicio de Endocrinología. Universidad Autónoma. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Introducción: En los últimos años, se han descubierto nuevos péptidos de producción adipocitaria con acciones endocrinas. Sus niveles plasmáticos se correlacionan con la cantidad, proporción y distribución de grasa corporal en el individuo. La modificación en la composición corporal que acontece a lo largo del desarrollo puberal, con características diferenciales entre ambos sexos, podría estar relacionada con cambios en la producción de citocinas adipocitarias.
Objetivos: 1. Establecer los valores de normalidad de adiponectina, resistina, factor de necrosis tumoral a (TNF-α) e interleucina 6 (IL-6) a lo largo del desarrollo. 2. Investigar la eventual existencia de dimorfismo sexual en los mismos. 3. Cuantificar las concentraciones circulantes del receptor soluble de leptina en esta población y evaluar su posible correlación con los de leptina y el resto de parámetros evaluados, así como con el índice de masa corporal (IMC).
Pacientes y métodos: Se estudiaron los niveles séricos de adiponectina, resistina, TNF-α, IL-6, leptina y su receptor soluble en 160 sujetos sanos (83 niñas y 77 niños) agrupados de acuerdo con su estadio de desarrollo puberal (Tanner I, 23 niñas y 22 niños; Tanner II, 19 niñas y 16 niños; Tanner III + IV, 21 niñas y 20 niños; Tanner V, 20 niñas y 19 niños). Las concentraciones de adiponectina y leptina de valoraron por radioinmunoensayo (Linco®, EE.UU.), las de resistina (Linco®, EE.UU), TNF-α e IL-6 (R&D Systems®, EE.UU.) y receptor soluble de leptina (BioVendor®, República Checa) mediante enzimoinmunoanálisis. En todos los casos, los coeficientes de variación intraensayo e interensayo fueron inferiores al 10 %.
Resultados: Los niveles de adiponectina presentan dimorfismo sexual a partir de la pubertad media (p < 0,001), debido a un descenso en varones (p < 0,05). La resistina no se modifica a lo largo del desarrollo, con excepción de las mujeres con pubertad finalizada (p < 0,001), que exhiben concentraciones circulantes elevadas. Las concentraciones de TNF-α no se modifican, mientras que las de IL-6 disminuyen significativamente en ambos sexos a partir de la media pubertad (p < 0,05). Cuando se analiza el cociente leptina/receptor soluble, se observa un incremento evolutivo, significativo sólo en mujeres (p < 0,001), que determina una diferencia entre sexos al finalizar la pubertad (p < 0,001). Las concentraciones circulantes del receptor soluble de leptina exhiben una correlación negativa con los de leptina (r = 0,27; p < 0,01), así como con el IMC (r = 0,32; p < 0,001) (tabla 1).
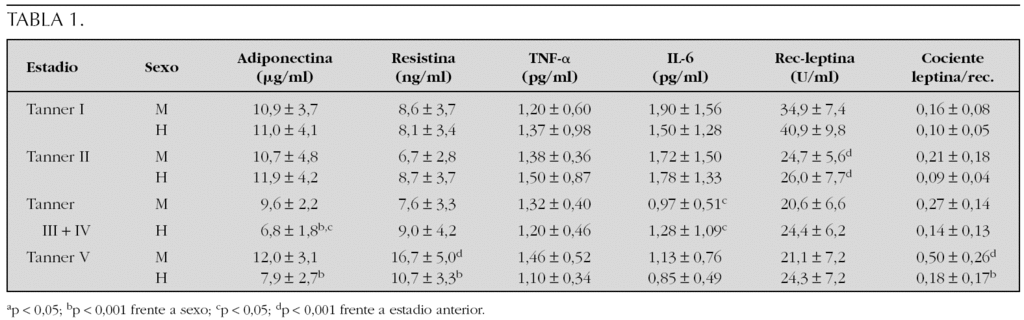
Conclusión: Los cambios de las adipocitocinas a lo largo del desarrollo puberal están en relación con las modificaciones en la composición corporal y el sexo.
Adipocitocinas y ghrelin en recién nacidos: relación con edad gestacional e índice ponderal
V. Barrios Sabador, G.A. Martos-Moreno, I. Sánchez, M. Sáenz-Pipaón, J. Quero y J. Argente
Endocrinología. Universidad Autónoma. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Introducción: La regulación de la homeostasis energética se ha estudiado en la infancia y adolescencia, desconociéndose los mecanismos que la modulan en las fases finales de la vida intrauterina. Recientemente, se han descubierto numerosos péptidos relacionadas con el equilibrio energético y la composición corporal, aunque su posible relación con el grado de maduración y nutrición del neonato permanece mal establecida.
Objetivos: 1. Determinar los niveles circulantes de adiponectina, resistina, leptina, receptor soluble de leptina, ghrelin total y acilado en recién nacidos. 2. Analizar la posible influencia del grado de maduración y nutrición sobre estos péptidos. 3. Investigar la posible correlación entre estos parámetros bioquímicos, así como con la edad gestacional y el índice ponderal.
Pacientes y métodos: Se estudiaron 46 recién nacidos pretérmino con una edad gestacional comprendida 26 y 36 semanas y 84 recién nacidos a término con una edad gestacional entre 37 y 41 semanas. Todos ellos nacieron tras embarazo controlado, sin incidencias y presentaban un peso adecuado a su edad gestacional. En todos ellos se calculó el índice ponderal (IP) mediante la fórmula gramos x 100/ml, y se distribuyeron en tres grupos: bajo (< 2,35), medio (2,35-2,65) y alto (> 2,65). Se determinaron en sangre de cordón los niveles de adiponectina, leptina, ghrelin total y acilado por RIA (Linco®, EE.UU.); resistina y receptor soluble de leptina por ELISA (Linco®, EE.UU. y BioVendor®, República Checa). En todos los ensayos, los coeficientes de variación intraensayo e interensayo fueron inferiores al 10 %.
Resultados: Los recién nacidos a término presentaron concentraciones de adiponectina y leptina significativamente superiores a los pretérmino (p < 0,01 y p < 0,001, respectivamente). Por el contrario, mostraron concentraciones de receptor de leptina, así como el índice ghrelin acilado/ghrelin total significativamente inferiores (p < 0,05). No se observaron diferencias significativas en las concentraciones séricas de resistina. No se encontraron diferencias entre los grupos establecidos según el IP en ninguno de los parámetros estudiados, ni en recién nacidos a término ni pretérmino. Existe una correlación positiva entre la edad gestacional y las concentraciones de adiponectina y leptina en sangre de cordón (r = 0,39; p < 0,001, y r = 0,47; p < 0,001, respectivamente), así como una relación inversa entre leptina y su receptor soluble (r = 0,27; p < 0,01). El índice ponderal muestra una correlación directa con las concentraciones de leptina (r = 0,36; p < 0,001) e inversa con los de su receptor soluble (r = 0,30; p < 0,01).

Conclusiones: 1. Los niveles de adipocitocinas y de la fracción de ghrelin acilado en el recién nacido son dependientes de su grado de maduración. 2. En ellos, las concentraciones de leptina y de su receptor soluble podrían ser buenos marcadores del estado nutricional.
Expresividad del hiperandrogenismo en portadores y formas crípticas genotipados de deficiencia de 21-hidroxilasa en relación con variantes polimórficas de los genes del receptor de andrógenos y la calpaina 10
B. Ezquieta Zubicaray, J.M. Bellón, R. Barrio, M. Gallego, A. Gutiérrez Macías, F. Hermoso, J.I. Labarta, J.L. Lechuga, J.P. López Siguero, L. Lorenzo, C. Luzuriaga, M. Oyarzábal, S. Quinteiro, A. Rodríguez, D. Rodríguez Arnao y S. Martínez
Laboratorio de Diagnóstico Molecular Bioquímica. Endocrinología Pediátrica. Hospital General Universitario Gregorio Marañón. H. Universitario Ramón y Cajal. H. 12 de Octubre. Madrid. H. Virgen del Camino. Navarra. H. Puerta del Mar. Cádiz. H. Materno Insular. Canarias. H. Marqués de Valdecilla. Santander. H. Virgen Arrixaca. Murcia. H. Universitario Valladolid. Madrid. España.
Introducción: El hiperandrogenismo es consecuencia de la interacción ambiental y poligénica de diversas variantes alélicas polimórficas existentes en la población normal. Los estudios previos, mayoritariamente en mujeres hiperandrogénicas, se han centrado en análisis de asociación monogénica con grupos control de comparación establecidos por criterios únicamente fenotípicos. El gen 21OH de forma recesiva monogénica da lugar a hiperandrogenismo y su alelo deficiente no clásico más frecuente V281L es muy prevalente en nuestra población. El hiperandrogenismo pediátrico se asocia con frecuencia con elevaciones leves de 17-hidroxiprogesterona tras ACTH.
Objetivo: Análisis de variantes polimórficas del receptor de andrógenos (AR) y calpaina 10, en hiperandrogenismo pediátrico (portadores y no portadores 21OHD) y formas crípticas familiares.
Casos y controles: 104 pacientes pediátricos con hiperandrogenismo (HF) y marcadores bioquímicos normales con excepción de la 17-hidroxiprogesterona tras ACTH, elevada en el rango definido para los portadores de 21OHD (4-12 ng/ml, Ezquieta et al, Acta Paediatr, 2002) 126 controles pediátricos (hermanos de casos índice clásicos y no clásicos), todos ellos evaluados por sus endocrinólogos pediatras, genotipados para el gen 21OH y clasificados como portadores (P, n = 83) y no portadores (NP, n = 43). Son 57 formas crípticas en familiares con diagnóstico molecular. Grupos HFP (n = 48), HFNP (n = 54), P, NP y Cr.
Estudio molecular: Genotipaje directo mediante técnicas de Southern y PCR/ASO para las mutaciones recurrentes del gen 21OH. Análisis indirecto de microsatélites: D6S273 y TNF-α y estudio de la expansión del receptor de andrógenos (AR) mediante PCR con primer fluorescente y separación en cromatografía capilar ABI 3100. Estudio de SNP por SnAPShot del polimorfismo UCSP-44 del gen de la calpaina 10. Análisis estadístico con SPSS 12.0, test exacto Fisher, χ 2 Pearson, significación p < 0,05.
Resultados: La distribución de alelos AR según el tamaño (n.º repeticiones CAG) de la expansión del exón 1 (poliGln) ha seguido un patrón similar al descrito en otras poblaciones (mediana 22 rep; 10-35). Cuando se compararon los portadores 21OH que habían presentado HF con aquellos que no, se encontró que los primeros presentaban alelos de tamaños menores (< 21rep Pc 25) con una frecuencia menor 4 frente a 28 % (p = 0,001). En concordancia con ello, y en el otro extremo del espectro clínico, las formas crípticas (mutaciones 21OH en ambos alelos) presentaban tamaños mayores de alelos (> 26, Pc75) con una menor frecuencia 12 frente a 31 % (p = 0,018). Se han considerado ambos alelos en XX o el único alelo X en varones. Con respecto al polimorfismo UCSNP-44 del gen de la calpaina 10, su distribución en conjunto 74 % TT/24,5 % TC/ 1,5 % CC se ajusta al descrito, con una distribución sesgada de la variante infrecuente, ya que los cuatro homozigotos detectados pertenecían al grupo HF (CC frente a CT + TT en HF frente a controles; p = 0,021). En lo que se refiere a la interacción de la variante C de UCSNP-44 y los alelos AR, tanto los homozigotos CC, todos HF, como los heterozigotos C/T con HF (n = 38) presentaban alelo/s > 22 rep.
Conclusiones: Los datos obtenidos no permiten confirmar que los alelos AR menores se asocien con una mayor expresividad del hiperandrogenismo pediátrico y, por el contrario, apuntan a que en el estricto rango de la normalidad pueda existir una relación directa expresividad/n.º CAG, sugiriendo quizá que en estos rangos pueda evidenciarse el efecto activador de transactivación ejercido por la poliGln descrito. Nuestros datos están de acuerdo con una asociación de la variante UCSNP-44 "C" descrita en mujeres OPQ por algunos autores, en este estudio asociada con el hiperandrogenismo pediátrico y siguiendo un modelo recesivo.
Estudio observacional en niñas con pubertad precoz central (PPC) tratadas con análogos de GnRH
L. Soriano Guillén, M.J. Martín-Díaz, B. Herrero-Mendoza, M. Gómez-Manchón, O. Rubio-Cabezas, M.T. Muñoz, J. Pozo y J. Argente
Endocrinología. Universidad Autónoma. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Antecedentes: El tratamiento con análogos de GnRH se ha mostrado eficaz en la detención del desarrollo de los caracteres sexuales secundarios en las niñas con PPC; sin embargo, los datos de talla adulta disponibles son contradictorios debido, al menos en parte, al método de predicción de talla utilizado. Además, los datos relacionados con la ganancia ponderal asociada son escasos.
Objetivos: 1. Analizar el porcentaje de casos de PPC idiopática y orgánica. 2. Estudiar los datos basales al diagnóstico. 3. Valorar la duración del tratamiento. 4. Comparar la talla adulta obtenida con la predicción de talla realizada al diagnóstico mediante el método de Bayley-Pinneau (average frente a accelerated). 5. Investigar el efecto de los análogos de GnRH sobre el peso e IMC. 6. Determinar el tiempo medio de aparición de la menarquia tras suspender análogos.
Sujetos y métodos: Se han estudiado retrospectivamente 27 pacientes afectadas de PPC, y se ha recogido lo siguiente: inicio de síntomas, patología de base, inicio de tratamiento, dosis de análogo, predicción de talla mediante el método de Bayley-Pinneau al diagnóstico, talla diana, duración del tratamiento y talla adulta. Así mismo, se han recogido los siguientes datos al diagnóstico y con una periodicidad anual: peso, talla, IMC, velocidad de crecimiento y edad ósea.
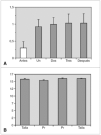
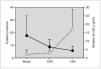
Resultados: La edad media al diagnóstico fue de 6,8 ± 1,6 años; la edad ósea de 8,9 ± 1,8 años; la velocidad de crecimiento (DE) de 3 ± 2,3 cm/año y el pico de LH de 24 ± 19 mU/ml. En ocho de ellas (32 %) existía patología orgánica. Se inició tratamiento con análogos de GnRH (dosis inicial media: 67 ± 17 μg/kg/ 28 días), durante un período de 3,9 ± 1,8 años. Durante el tratamiento, se objetivó un incremento significativo del IMC (fig. 1a) que se mantuvo tras la retirada de los análogos. La talla adulta obtenida fue de 158,2 ± 7,7 cm, sin presentar diferencias significativas ni con la talla diana ni con la predicción de talla al diagnóstico según los dos modelos utilizados (fig. 1b); sin embargo, aunque de manera no significativa, la talla adulta fue superior en 4 cm con respecto al pronóstico average. Finalmente, el tiempo medio de aparición de la menarquia tras la retirada de los análogos fue de 14 ± 4,2 meses.

Figura 1.
Conclusiones: 1. El tratamiento con análogos de GnRH es eficaz en las pacientes con PPC, alcanzando una talla adulta acorde con la talla diana. 2. La utilización del método de predicción de talla por Bayley-Pinneau mediante la edad media parece más adecuado, con el objeto de evitar una posible supraestimación en la talla adulta si se utiliza el modelo de edad ósea acelerada. 3. La administración de análogos incrementa el IMC, por lo que es necesario recomendar medidas higiénico-dietéticas durante dicho tratamiento.
Estudio observacional de niños con pubertad precoz central (PPC) tratados con análogos de GnRH
L. Soriano Guillén, M.J. Martín-Díaz, O. Rubio-Cabezas, M.T. Muñoz, J. Pozo y J. Argente
Endocrinología. Universidad Autónoma. Hospital Infantil Universitario Niño Jesús. Madrid. España.
Introducción: La prevalencia de pubertad precoz central (PPC) es menor en niños que en niñas, pero con mayor incidencia de patología orgánica. Igual que en las niñas, el tratamiento con análogos de GnRH es eficaz para frenar el desarrollo puberal; sin embargo, existe una enorme controversia sobre la talla adulta (TA) alcanzada y su diferencia con los diversos métodos de predicción de talla.
Objetivos: 1. Conocer el porcentaje de PPC secundaria a patología orgánica en niños. 2. Comparar la talla adulta obtenida con la predicción de talla realizada al diagnóstico mediante método de Bayley-Pinneau (Average frente a Accelerated). 3. Estudiar el efecto de los análogos de GnRH sobre peso e índice de masa corporal. 4. Determinar si los niveles de testosterona son un buen indicador de la eficacia terapéutica. 5. Investigar la velocidad de crecimiento (VC) previa al diagnóstico y durante el tratamiento.
Sujetos y métodos: Se han estudiado retrospectivamente 8 pacientes varones afectados de PPC, recogiendo los siguientes datos clínicos: inicio de síntomas, patología de base, inicio de tratamiento, dosis de análogos de GnRH, predicción de talla mediante método de Bayley-Pinneau al diagnóstico, talla diana, duración del tratamiento y TA. Además, se determinó al diagnóstico y con una periodicidad anual: peso, talla, IMC, velocidad de crecimiento, edad ósea y niveles de testosterona.
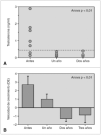
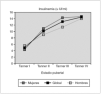
Resultados: La edad media al diagnóstico fue de 5,9 ± 2,9 años; la edad ósea de 7 ± 3 años; la velocidad de crecimiento (DE) de 2,2 ± 1,9 cm/año; el pico de LH de 12,9 ± 6,5 mU/ml y el volumen testicular de 6,1 ± 3,1 ml (rango: 4-12 ml). Del total, 6 niños presentaban patología orgánica (75 %): un hamartoma hipotalámico, un paciente presentaba secuelas de un traumatismo craneoencefálico grave, 2 pacientes con neurofibromatosis tipo I (ambos con gliomas del nervio y del quiasma óptico), un quiste supraselar con ventriculomegalia y un astrocitoma hipotálamo-quiasmático. La dosis inicial de tratamiento con análogos de GnRH fue de 65 ± 6 μg/kg/28 días, y se mantuvo durante un período de 6 ± 3,4 años, ajustándose periódicamente al peso. Durante el tratamiento, 7 pacientes mostraron un test LHRH frenado a los 3 meses de iniciado el tratamiento. Un único paciente no cumplió adecuadamente las indicaciones terapéuticas, y a los 3 meses presentó un pico de LH superior a 10 junto a niveles de testosterona de 3 ng/ml (VN < 0,5 ng/ml), mientras el resto presentó concentraciones por debajo de 0,5 ng/ml durante el tratamiento (fig. 1a). Así mismo, tras la instauración de análogos de GnRH, se evidenció una disminución significativa de la velocidad de crecimiento (fig. 1b), sin evidenciar cambios en la diferencia entre la edad ósea menos la edad cronológica durante el tratamiento, debido a la escasa diferencia existente al diagnóstico (1,1 ± 2,4). Durante la terapia con análogos de GnRH no hubo alteraciones del peso ni del IMC. Aunque sólo 3 pacientes han alcanzado la TA, esta fue de 170 ± 1,4 cm, sin presentar diferencias significativas ni con la talla diana ni con la predicción de talla al diagnóstico según el modelo average, pero sí con el modelo accelerated (8 cm).
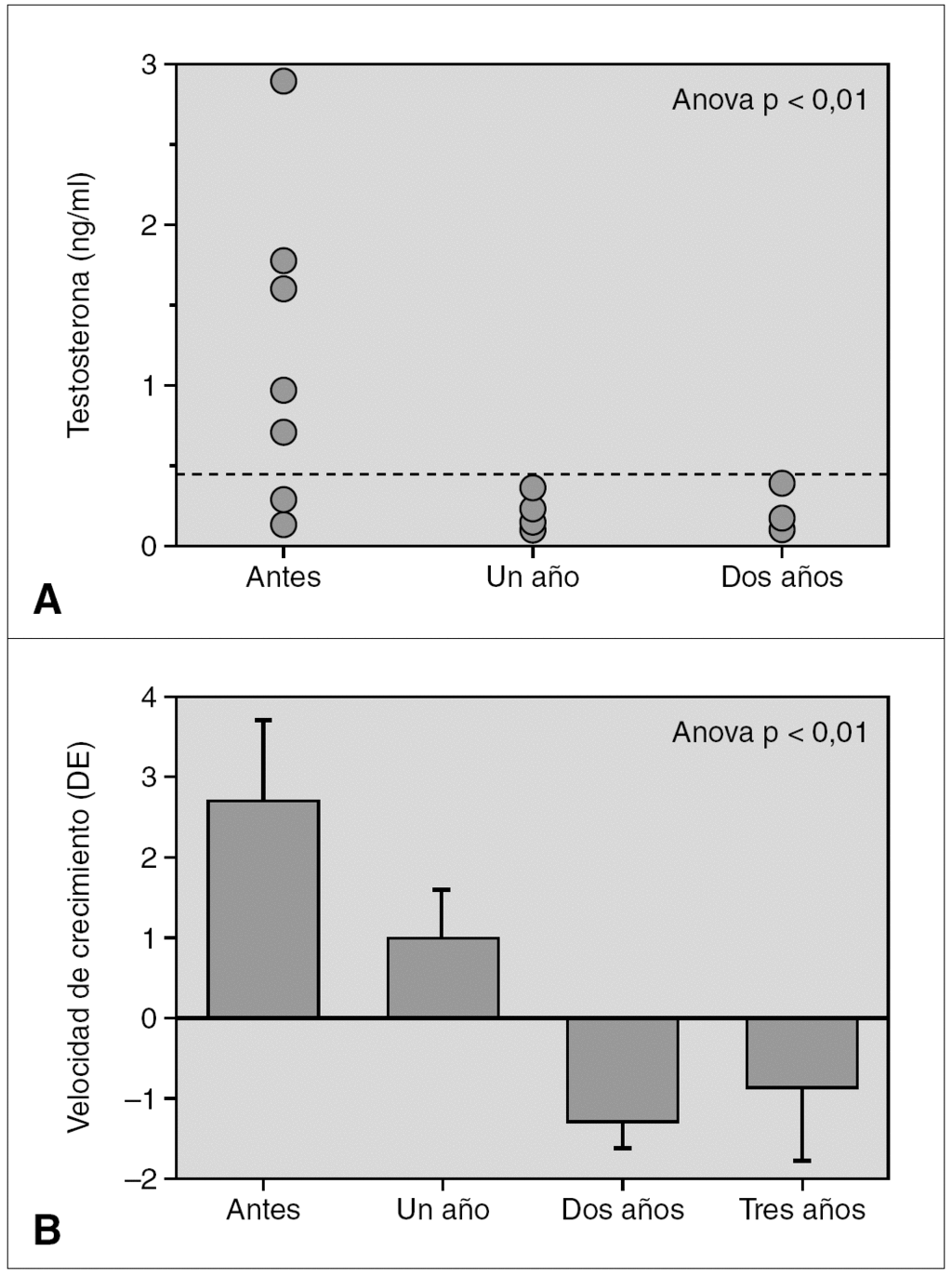
Figura 1.
Conclusiones: 1. La presencia de PPC en niños obliga a descartar patología orgánica central. 2. La utilización del método de predicción de talla por Bayley-Pinneau en niños presenta más errores respecto a la TA que en niñas con esta patología, sobre todo si se utiliza el modelo de edad ósea acelerada. 3. La determinación de la concentración de testosterona podría ser un excelente parámetro para evaluar la eficacia terapéutica de los análogos de GnRH, pudiendo sustituir al test de LHRH.
CRECIMIENTO
Caracterización auxológica de los nacidos PEG
M. Martín Alonso, L.M. Prieto Tato, B. Nieto Almeida, E. Álvarez Aparicio, J. Cedeño Montaño y J. Prieto Veiga
Unidad de Endocrinología Pediátrica. Hospital Clínico Universitario de Salamanca. España.
Introducción: Los niños que nacen pequeños para la EG (PEG) tienen longitud y/o peso < 2 DE de las curvas de referencia. Ambos parámetros y el perímetro cefálico (PC) se afectan de distinta manera según la causa y el momento del deterioro pudiendo ser así el niño proporcionado o especialmente delgado.
Objetivos: 1. Análisis auxológico de un grupo de PEG de nuestro Hospital. 2. Evaluación de la frecuencia e intensidad con que se deterioran peso, longitud y PC. 3. Valoración del grado de nutrición fetal. 4. Ayudar al conocimiento del crecimiento recuperador del grupo. 5. Establecer las bases de un estudio clínico-bioquímico ya iniciado.
Casuística: Grupo de 60 niños PEG a los que se hace seguimiento de parámetros auxológicos y bioquímicos. Se han excluído gestaciones múltiples y cuadros sindrómicos. EG: 35-41 sem. EC > 2 años. Sexo: 21 V, 39 M.
Metodología: Primera evaluación en la Unidad Neonatal. Seguimiento en consulta externa. Variables: EG, sexo, peso, longitud, PC e índice ponderal (IP) en DE utilizando como referencia las tablas de crecimiento fetal de Carrascosa (2003).
Resultados: Por definición, el peso fue < 2DE en todos los casos. La longitud fue < 2DE en el 28 % en ambos sexos y el PC en el 18-19 %. El descenso del IP fue más frecuente entre las niñas (43,58 %) pero la intensidad algo superior en varones (1,95 DE) (tabla 1).

Conclusiones: 1. Mayor frecuencia de PEG en niñas. 2. No encontramos diferencias cualitativas ni cuantitativas entre ambos sexos que sean significativas. 3. El carácter asimétrico de la mayoría de los casos, hace pensar en un origen tardío del problema. 4. El seguimiento clínico de estos pacientes puede aportarles beneficios a la hora de establecer un pronóstico futuro en cuanto a calidad de vida y salud.
Estudio de eficacia y seguridad de Genotonorm®en SGA
R.M. Espadero Amat, A. Carrascosa y Grupo Colaborador Español
Unidad de Endocrinología Pediátrica. Unidad Médica, Pfizer España y Hospital Infantil Vall d'Hebron. Alcobendas. Madrid. España.
Objetivo: Estudiar la eficacia de la rhGH sobre el crecimiento (SDE de la talla) en pacientes de talla baja con antecedentes de RCI comparándolo con un grupo no tratado. Evaluar la seguridad del tratamiento con Genotonorm® en los pacientes en tratamiento.
Material y métodos: Un total de 132 pacientes (67 niños, 65 niñas) prepuberales. Pacientes tratados n = 66 (grupo TTO), no tratados n = 66 (grupo NTTO). Criterios de inclusión I: peso y/o talla al nacer < p10 (Delgado, 1996), con una edad gestacional > 35 semanas, edad cronológica > 3 años, SDE de talla < -1,88 (Hernández, 1988), VC < P50 para E.C. (Hernández, 1988), cariotipo normal en niñas, respuesta de GH ≥ 10 ng/ml. Los padres debían dar su consentimiento informado. Tratamiento: genotonorm a una dosis: 66 μg/kg/día inyección subcutánea diaria nocturna durante 2 años en grupo tratado.
Resultados (12 meses): No hay diferencias estadísticas entre grupos en peso y longitud al nacer, edad gestacional, tallas de los padres, ni talla diana. La EC en la visita basal fue de 7,7 ± 2,7 años en el grupo TTO frente a 7,3 ± 2,5 años en NTTO. Se resumen los resultados en la siguiente tabla 1.
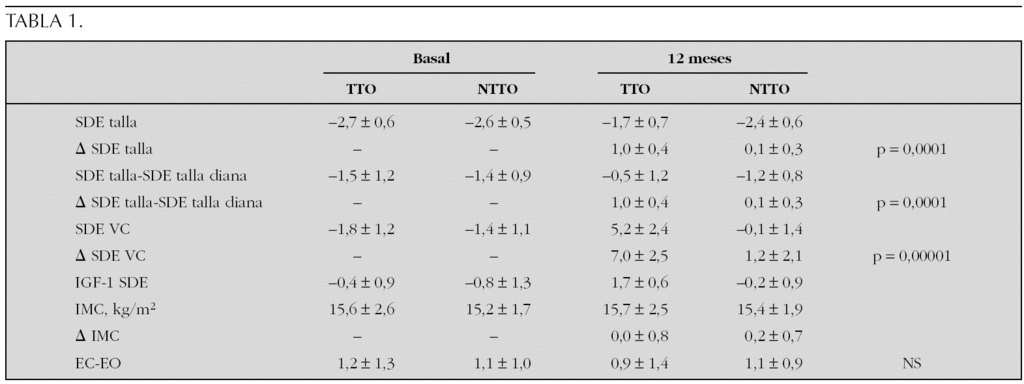
En cuanto a sucesos adversos (SA), hubo un total de 101 (75 en el grupo de TTO y 25 en el grupo de NTTO) en 57 pacientes (39 pacientes en TTO y 18 pacientes en NTTO). De estos SA dos fueron graves (uno en grupo TTO, uno en grupo NTTO), siendo en el grupo tratado una epifisiólisis de cabeza femoral y una apendicitis aguda en el grupo NTTO. SA de tipo metabólico: se dieron en el grupo TTO 7 SA (tres hiperinsulinemias, dos ↑ HbA1c, una ↑ IGF-I, una hipertrigliceridemia, una ↑ transaminasas); en el grupo NTTO 5 SA (una ↓ IGF-I, una ↓ IGFBP-3, un hipotiroidismo subclínico, hipercolesterolemia, una hipertrigliceridemia).
Conclusiones: El tratamiento con GH en los pacientes con antecedentes de RCIU mejora la talla significativamente acercando a los pacientes a su talla diana. A lo largo de 12 meses de seguimiento no hay variación del IMC en ninguno de los dos grupos.
Retraso del crecimiento intrauterino asociado a patología infrecuente
M.M. Martínez López, M. Álvarez-Acevedo, I. González Casado, E. Martín Campagne, J. Solera García y R. Gracia Bouthelier
Servicio de Endocrinología Pediátrica. Hospital Infantil La Paz. Madrid. España.
Introducción: El retraso del crecimiento intrauterino (RCIU) representa un grupo heterogéneo de pacientes caracterizados por antropometría neonatal insuficiente para su edad gestacional. La regulación del crecimiento prenatal es compleja y no totalmente conocida; existen distintos factores causales que pueden originar el RCIU. Cada vez cobra mayor importancia el complejo control hormonal del crecimiento fetal, con el papel determinante de la IGF-1 y la insulina. Es conocida la relación entre el déficit insulínico, las cifras bajas de IGF-1 y la falta de recuperación del crecimiento posnatal. Presentamos un caso complejo de un niño con RCIU asociado a patología digestiva y a presentación inusual de diabetes mellitus, en el que no existe recuperación de talla posnatal.
Caso clínico: Niño con RCIU (peso de recién nacido < 1 DE, talla < 2 DE) estudiado desde los primeros días de vida en el Servicio de Gastroenterología por malabsorción de glucosa-galactosa, trastorno motor del colon y esteatorrea. En tratamiento con dieta hipercalórica, hipergrasa, pobre en glucosa y sin lactosa. No recuperación ponderoestatural, permaneciendo por debajo del percentil 3 desde el nacimiento. Es derivado al Servicio de Endocrinología a los 8 años de edad por presentar, a pesar de la malabsorción de glucosa-galactosa, cifras de diabetes en la SOG. Exploración física: Peso 22,7 kg, talla 113 cm (2,4 DE), armónico, no dismorfias, exploración general normal. Antecedentes familiares: Madre diagnosticada de DM tipo 1 desde los 17 años, tía paterna con diabetes gestacional, abuelo paterno y tía abuela paterna con DM tipo 2 en tratamiento con antidiabéticos orales y tío abuelo paterno con DM tipo 2 e insulinoterapia. Exploraciones complementarias: SOG: glucemia basal de 117 mg/dl, a los 120 min de 337 mg/dl, insulinemia basal de 4 μU/ml, a los 120 min de 20 μU/ml; péptido C basal de 0,5 ng/ml, tras glucagón: 3,9 ng/ml; anticuerpos anti-GAD, anti-IA2 y antiinsulina negativos; IGF-1: 75 ng/ml, IGF-BP3: 2,7 μg/ml; GH con hipoglucemia insulínica: basal 1 ng/ml, pico 4 ng/ml; GH propranolol-ejercicio: basal 2,5 ng/ml, pico 9 ng/ml; edad ósea de 5 años. Con estos datos no parece probable DM tipo 1 o 2 y se sospecha una diabetes tipo MODY, solicitándose estudio genético. El estudio molecular permitió identificar la inserción 153 ins TGGGGGT en la región promotora del gen HNF-1α, mientras que el resto del gen era rigurosamente normal. Esta alteración fue identificada tanto en el paciente como en su madre. La inserción podría afectar el normal funcionamiento de la actividad promotora. Esta mutación ha sido previamente identificada en pacientes noruegos con un fenotipo clínico también compatible con diabetes MODY tipo 3 y en los que el resto del gen era así mismo normal. Evolución: Inicialmente se instaura tratamiento dietético y ejercicio físico reglado, con buen control de HbA1c. Con 10 años y medio se inicia tratamiento con antidiabéticos orales por empeoramiento del control metabólico y de HbA1c (7 %). Se solicita protocolo de tratamiento con GH para RCIU.
Discusión: Se trata de un caso complejo de asociación de RCIU con malabsorción de glucosa-galactosa y diabetes MODY tipo 3, en el que se desconoce el papel etiopatogénico que puede desempeñar la alteración del HNF-1a. El retraso de talla es posiblemente multifactorial y podría beneficiarse del tratamiento con GH.
Secreción de melatonina por la glándula pineal, durante test de estímulo de hormona de crecimiento
E. Caro Cruz, J.M. Fernández-García, C. Ruiz-Cosano y A. Muñoz-Hoyos
Pediatría. Complejo Hospitalario de Jaén. España.
Introducción: Existen estudios experimentales sobre las interrelaciones de la hormona de crecimiento y la melatonina. Así se ha observado que en ratas ciegas o mantenidas en oscuridad constante y prolongada reducen su peso y la longitud de la tibia y su hipófisis contiene menos hormona de crecimiento con relación a un grupo control. Existen trabajos realizados en seres humanos, adultos, en los que existen resultados contradictorios. Así Wright et al no han encontrado efecto alguno de la melatonina sobre los niveles plasmáticos de LH, GH, T4, testosterona y cortisol. Lissoni et al demostraron una disminución de las concentraciones plasmáticas de GH después de la administración aguda de melatonina en niños prepuberales y finalmente Valcavi et al. Observaron que existe un incremento de la GH basal tras la administración oral de melatonina y que ésta disminuye la respuesta de hormona de crecimiento tras hipoglucemia insulínica.
Objetivos: Intentamos estudiar la existencia de interrelación entre hormona de crecimiento y melatonina en niños y si existe de qué tipo.
Material y métodos: Se han determinado las tasas plasmáticas de la hormona de crecimiento en 3 grupos de niños, todos ellos con el diagnóstico de baja talla y que fueron sometidos a tests de estímulo de hormona de crecimiento con: propranolol + ejercicio, clonidina e hipoglucemia insulínica. Se estudiaron para cada test, grupos de 34, 31 y 30 niños, respectivamente. Las determinaciones de hormona de crecimiento y melatonina se realizaron mediante radioinmunoanálisis.
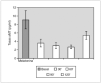
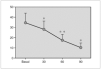
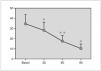
Resultados: Pudimos observar en los tres grupos de pacientes una relación inversa entre la secreción de la hormona de crecimiento y melatonina, como se aprecian en las siguientes figuras 1, 2 y 3.
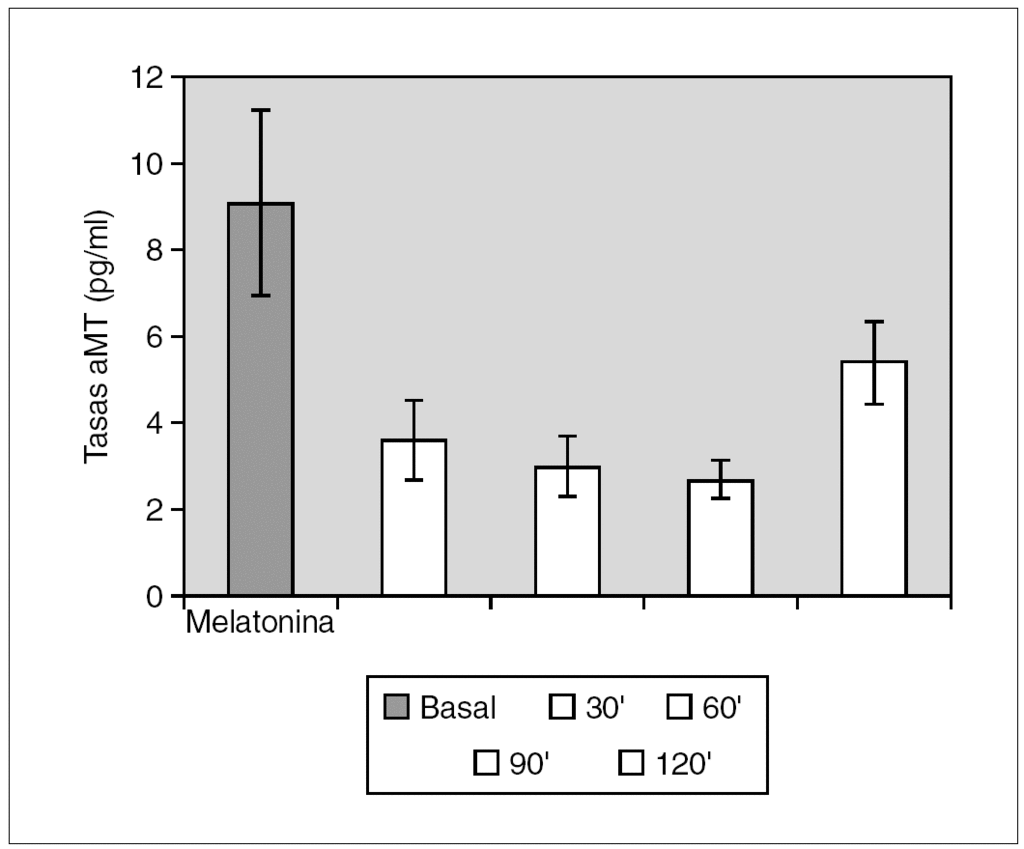
Figura 1. Secreción de melatonina tras un estímulo agudo. Hipoglucemia insulínica.
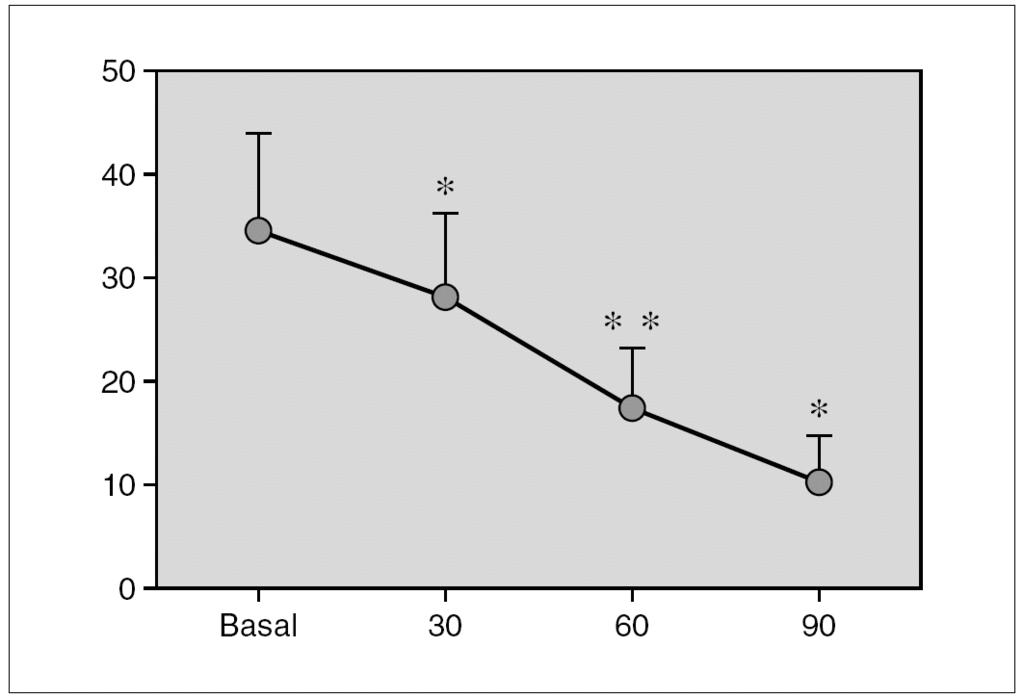
Figura 2.
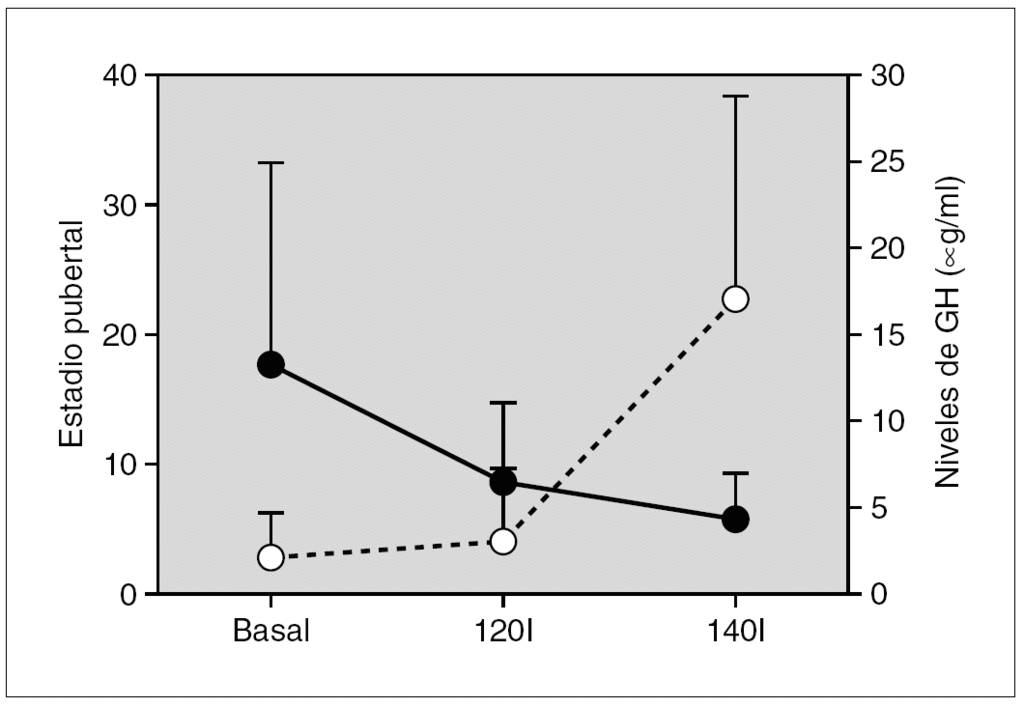
Figura 3.
Discusión: Los resultados que se muestran indican que en nuestros respectivos grupos la respuesta de la melatonina a un estímulo agudo sobre la secreción hipofisaria de la hormona de crecimiento es inversa a la secreción de la hormona de crecimiento. La actuación de los receptores pineales es distinta según se considere un estímulo o estrés agudo o crónico. En nuestro grupo hemos demostrado cómo una situación de estrés crónico se acompaña de alteraciones en la secreción de melatonina.
Efecto del tratamiento con hormona de crecimiento sobre el metabolismo hidrocarbonado y la resistencia insulínica en niñas con síndrome de Turner
C. Luzuriaga Tomás, M.ªC. García Gómez y J.L. Guerra Díez
Servicio de Endocrinología Infantil. Hospital Universitario Marqués de Valdecilla. Santander. España.
Introducción: Se ha descrito asociación entre el ST y alteración del metabolismo hidrocarbonado (M HdC) y si el tratamiento con hormona de crecimiento (GH) aumenta el riesgo de padecer diabetes mellitus tipo 2 o tolerancia deteriorada a hidratos de carbono (T HdC).
Objetivo: Comprobar si existe patología inicial de alteración MH y analizar la repercusión del tratamiento con GH.
Método: En 19 pacientes ST en los que se ha realizado sobrecarga de glucosa oral 75 mg/m 2 s.c.; se han estudiado niveles de glucosa, insulina y péptido C. Área bajo la curva (ABC) de glucosa e insulina, criterios de insulinorresistencia: HOMA y Quicki e índice insulinogénico antes de iniciar el tratamiento con GH (dosis 0,42 mg/kg/día), durante el mismo (n = 17) a los 16-18 meses. En 6 casos después de retirar, mínimo 6 meses, la GH, todas las pacientes han recibido tratamiento dietético evitando hidratos de carbono de absorción rápida.
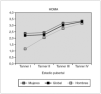
Resultados: véase tabla 1. El análisis estadístico se ha realizado con el paquete estadístico SPSS versión 11.0. Sólo una paciente presentó tolerancia deteriorada a hidratos de carbono según criterios ADA 1997 previo al tratamiento con GH, valor de glucosa a los 120 min:143 mg/dl. La HbA1c ha sido en todos los controles pre-GH y post-GH normales. Ninguna paciente mostró tolerancia deteriorada a los hidratos de carbono durante tratamiento con GH ni tras suprimirlo. En el análisis de relación de datos pre-GH y post-GH resultó: no relación estadística entre la glucosa basal, la glucosa a los 120 min, el área bajo la curva de glucosa y Quicki; casi significativa los niveles insulina basal (p = 0,074), HOMA (p = 0,074). Ha resultado significativa, el valor de insulina 120 min (p > 0,05), el área bajo la curva de la insulina (p < 0,05) y el valor del péptido C. El índice insulinicogénico pre-GH y durante el tratamiento con GH, aunque se incrementó no fue significativo 0,32 frente a 0,94 (p = 0,066). Los estudios realizados tras retirar la GH (n = 6) dio como resultado glucosa basal y a los 120 min similar (76,7 ± 7,6, 104,3 ± 11,7 mg/dl, respectivamente), no significativa el área bajo la curva de glucosa (19.290 ± 1.985). La secreción de insulina fue similar (10,2 ± 9,9) y el área bajo la curva de insulina fue inferior (7.823 ± 5.453). El péptido C aumentó 2,2 ± 0,9; desciende el HOMA de 3,1 a 2,6 y aumenta el Quicki a 0,36. La comparación de datos de los pacientes, tras retirar GH, no ha sido significativa comparando con los valores iniciales y durante el tratamiento con GH.
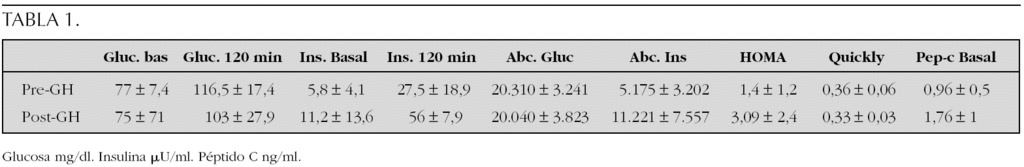
Conclusiones: Según nuestros datos, en las pacientes ST durante el tratamiento con GH es evidente un incremento de la secreción de insulina y péptido C sin que llegue a observarse una mayor insulinorresistencia o una disminución importante de la sensibilidad a la insulina. Esta secreción de insulina aumentada evita la hiperglucemia. Aunque es escaso el número de pacientes estudiadas tras retirar GH, el hiperinsulinismo se corrige o mejora. El hecho de haber encontrado una tolerancia deteriorada a hidratos de carbono previo tratamiento con GH nos aconseja que debemos estudiar el metabolismo de la glucosa previo al tratamiento con GH. Ha podido tener importancia en nuestras pacientes, el seguimiento del metabolismo de hidratos de carbono y el tratamiento dietético realizado durante el tratamiento con GH y al suprimirlo.
Uso racional de las pruebas farmacológicas en el estudio de la reserva de GH en población infantil
M. Zapico Álvarez-Cascos, L. Ruiz Pérez, S. Lorenzo, E. Rodríguez, M. Mauri, R. Alfayate, A. López, P. Pérez Yuste, L. Santos y A. Pico
Unidad de Endocrinología Infantil. Unidad de Hormonas. Hospital General Universitario de Alicante. España.
Introducción: El estudio de la reserva de hormona del crecimiento (GH) en niños con baja talla continúa siendo un tema controvertido, no sólo por la pobre reproducibilidad de las pruebas dinámicas disponibles, sino también por la inexistencia de una prueba bioquímica de referencia.
Objetivos: Establecer la tasa de respuesta a test hipoglucemia-insulínica (ITT) como primera prueba y test de clonidina (CL) para diagnóstico de deficiencia de GH. Determinar el coste, en URC (Unidades Relativas de Coste), de ambas pruebas.
Material y métodos: Estudio retrospectivo en 87 niños (38 niñas y 49 niños) con edades comprendidas entre los 2 y los 12 años. Todos ellos fueron sometidos a la prueba de ITT por sospecha clínica de deficiencia de GH (determinación de cortisol y GH a 15 min y basal, administración de 0,10 U/kg de insulina rápida y determinación de glucosa, cortisol y GH a 15, 30, 45, 60 y 90 min). En aquellos en los que no se obtuvo respuesta (pico GH < 10 ng/ml), se realizó la prueba de CL (determinación basal de GH, administración oral de clonidina 0,075 mg/m 2 de superficie corporal en menores de 10 años o 0,15 mg/m 2 en mayores de 10 años, y determinación de GH a 30, 60, 90 y 120 min) y se considera que no respondieron a esta última, aquellos pacientes con un pico de GH < 10 ng/ml. La GH fue determinada mediante inmunoanálisis de quimioluminiscencia en IMMULITE 2000© de DPC y la IGF-1 por IRMA (Immunotech). Las URC vienen definidas y se calculan según el Catálogo del Sistema de Información Económica de la Conselleria de Sanitat de la Generalitat Valenciana.
Resultados: De los 87 niños, 20 (23 %) respondieron a ITT (pico respuesta GH 14,96 ± 7,22 ng/ml) mientras que 67 (77 %) no lo hicieron (pico respuesta GH 3,71 ± 2,5 ng/ml). De éstos, 28 (41 %) respondieron a CL mientras que 39 (58 %) no lo hicieron. En cualquier caso, tanto en respondedores como en no respondedores, la respuesta a GH fue superior en CL que en ITT (14,17 ± 4,33 frente a 5,47 ± 2,03 ng/ml en respondedores y 3,15 ± 2,9 frente a 2,87 ± 2,6 ng/ml en no respondedores). El coste, en URC, de una ITT es de 460,15 frente a 278,69 de una CL. Considerando el número de pacientes que ha respondido a CL y no a ITT (desconociendo el número de respondedores a ITT que no responden a CL), si CL hubiera sido la única prueba realizada, el ahorro hubiera sido de 10.887,6 URC.
Conclusiones: La prueba de la clonidina ofrece una mejor tasa de respuesta, es más económica y es mejor tolerada que ITT. La ITT ofrece la ventaja, frente a clonidina, de explorar la función corticosuprarrenal, pero determinaciones basales de cortisol pueden dar información suficiente en la mayoría de casos.
Test de hipoglucemia insulínica: discordancia en la respuesta de GH y cortisol con relación a la recuperación de la hipoglucemia de forma espontánea o con la administración de una solución glucosada
R. Tomasini, G. Dodino, D. Yeste, N. Potau, E. Armengol, M. Losada, M. Gussinyé y A. Carrascosa
Servicio de Endocrinología Pediátrica. Hospital Materno Infantil Vall d'Hebron. Barcelona. España.
Introducción: El test de hipoglucemia insulínica (THI) se considera el estándar de referencia y de elección para el diagnóstico de las alteraciones del eje hipotálamo-hipófiso-adrenal. En la literatura médica existe controversia sobre la conveniencia de recuperar la hipoglucemia inducida por la insulina, ya que ésta no está exenta de riesgos potencialmente graves.
Objetivo: Conocer si la recuperación de la hipoglucemia inducida por la insulina con una solución glucosada determina una diferente respuesta de la secreción de GH y de cortisol en los distintos tiempos del estudio con relación a la recuperación espontánea de la hipoglucemia por el paciente.
Sujetos y métodos: Estudio prospectivo de una población de 100 niños y adolescentes afectados de retraso de crecimiento y a los que se realizó un test de THI (dosis: 0,1 U/kg peso IV). En 50 pacientes la recuperación de la glucemia se produjo de forma espontánea y en el resto la recuperación se realizó con solución glucosada por vía oral (15-20 g de glucosa) cuando las glucemias fueron inferiores a 30 mg/dl (tabla 1; valores expresados como media y DE).

No se encontraron diferencias estadísticamente significativas entre los 2 grupos estudiados.
Resultados: En la tabla 2 se muestra la respuesta de la secreción de GH y de cortisol en todos los tiempos evaluados en las dos poblaciones y la significación estadística.

El déficit de GH se diagnosticó en 47 de los 50 pacientes recuperados con solución glucosada (94 %) y en 34 de los que recuperaron espontáneamente la hipoglucemia (68 %).
Conclusiones: La recuperación de la hipoglucemia con solución glucosada durante el THI determina una menor respuesta de GH y cortisol al estímulo de la insulina y una mayor frecuencia de diagnósticos de déficit de GH.
Pamidronato y GH en trasplantado de medula ósea
R. López Almaraz, L. Martín Viota, A. Concepción García, A. Sancho Pascual, J.C. Rodríguez Luis y J.P. González Díaz
Servicio de Pediatría. Hospital Universitario de Canarias. La Laguna. Tenerife. España.
Introducción: La inmunosupresión, la irradiación corporal total y la inmovilización prolongada contribuyen a la pérdida de masa ósea en pacientes postrasplantados de médula ósea (TMO). La utilización de bifosfonatos como inhibidores de la resorción ósea, si bien es habitual en pacientes adultos oncológicos y/o con osteoporosis, en la edad pediátrica es aún limitada. Presentamos el caso de un paciente post-TMO, tratado con pamidronato sódico intravenoso, y sus beneficios al tratamiento de la osteoporosis secundaria grave que padece.
Observación clínica: Varón de 12 años y 10 meses, trasplantado hace 16 meses de médula ósea (alogénico de hermana histocompatible) tras recaída precoz de leucemia linfoblástica aguda (LLA) estirpe B, fenotipo común. Complicaciones post-TMO: EICH crónica cutánea grado III e intestinal grado II, alteración del patrón de crecimiento (velocidad de crecimiento post-TMO = 0,5 cm/año, con pico de GH máximo tras test de hipoglucemia insulínica = 4,34 ng/ml), dolores osteomusculares intensos en rodillas y región lumbosacro con aplastamiento vertebral. Desde el trasplante está en tratamiento inmunosupresor con metilprednisolona y ciclosporina A. La densitometría ósea (DMO) de columna lumbar (L1-L4) evidenció una pérdida de densidad mineral ósea del 46 % (0,448 g/cm 2, z-score de 3,9 DE). Se decide iniciar tratamiento con pamidronato disódico intravenoso (30 mg/mes durante 3 meses y posteriormente 30 mg cada 3 meses) junto con suplemento de calcio oral (1 g/día) y vitamina D (400 U/día). Los marcadores óseos de formación (FAO y osteocalcina) y de resorción (calciuria y D-piridinolina) se encontraban al inicio alterados y mejoraron significativamente a lo largo del tratamiento, así como la DMO (0,532 g/cm 2, z-score de 3,32 DE, con una ganancia del 18,8 % a los 6 meses), desaparición casi total de los dolores osteomusculares y mejoría de la deambulación. No han aparecido efectos secundarios durante el tratamiento. A los 15 años (3 años y 5 meses post-TMO), persiste gran alteración del patrón de crecimiento: talla 3 DE, velocidad de crecimiento < 1,5 cm/año, con pico máximo de GH tras estímulo con clonidina = 6,1 ng/ml, IGF-1 = 405 ng/ml, IGFBP-3 = 4,32 mg/l, con una EO = 12 años y ausencia de signos puberales, motivo por el que se inició tratamiento con rhGH, mejorando la calidad de vida, la DMO (aumento del 18 %) e inició el desarrollo puberal.
Patrones antropométricos de los recién nacidos a término de población inmigrante de raza negra, árabe e hispanoamericanos (n = 2.444)
A. Copil 1, D. Yeste 1, R Teixidó 2, J. Maciá 3, M. Gussinyé 1 y A. Carrascosa 1
1Servicio de Endocrinología Pediátrica. Hospital Materno-Infantil Vall d'Hebron. Barcelona. 2Hospital de Olot. Girona. 3Hospital Josep Trueta. Girona. España.
Introducción: El incremento de la inmigración en las últimas décadas en España ha determinado un aumento del número de recién nacidos de raza no caucásica en nuestro medio.
Objetivo: Establecer valores de normalidad de desarrollo intrauterino del peso (g), longitud (cm) y perímetro craneal (cm) de una población de recién nacidos a término (RNAT) producto de gestaciones únicas, sin malformaciones congénitas y de curso normal de raza negra (África subsahariana), árabe (Marruecos) e hispanoamericanos (Ecuador y Perú).
Sujetos y métodos: Se han incluido un total de 2.444 RN de 37-42 semanas de gestación (1.230 varones/1.214 mujeres) de las siguientes razas: negra (1.257 RNAT, 622 niños/635 niñas), árabe (520 RNAT, 297 niños/223 niñas) e hispanoamericanos (667 RNAT, 356 niños/311 niñas).
Resultados: En la tabla se muestra para cada edad gestacional y en función del sexo los valores expresados como media y DE del peso (g), longitud (cm) y perímetro craneal (cm) de los RNAT de cada grupo étnico (tabla 1).
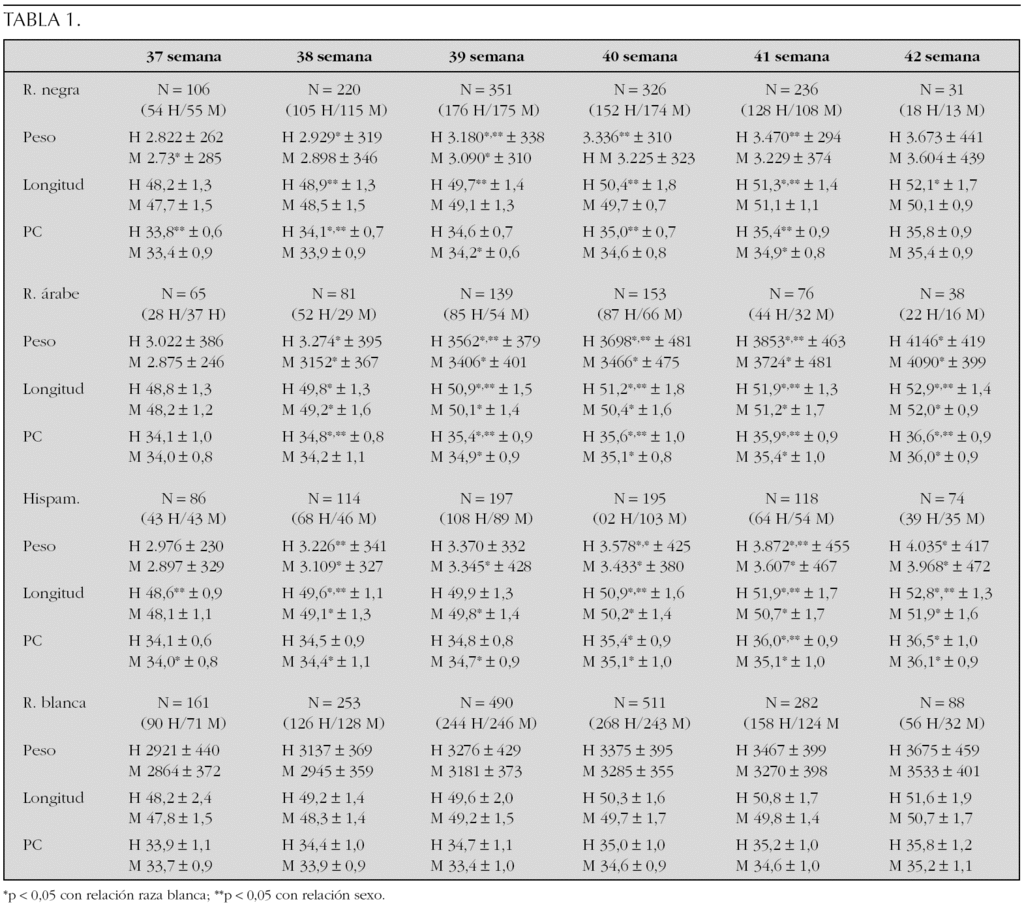
Conclusiones: La existencia de dimorfismo sexual en los parámetros antropométricos evaluados no es constante en todas las edades gestacionales consideradas. Los patrones antropométricos de la población de RNAT de raza caucásica y nacionalidad española no son extrapolables a la hora de evaluar un RNAT de otra etnia.
Raquitismo hipofosfatémico familiar
J.P. González Díaz, A. Concepción, A. Sancho, L. Martín Viota y R. López Almaraz
Servicio de Pediatría. Hospital Universitario de Canarias. Tenerife. España.
Introducción: La similitud clínica entre muchas displasias esqueléticas ha hecho difícil, en muchas ocasiones, el diagnóstico definitivo, como ocurrió en la familia de nuestra paciente, que en la rama materna, hasta una tercera generación presentó alteraciones esqueléticas en la infancia, que fueron etiquetadas de displasia metafisaria de tipo Jansen.
Observación clínica: Mujer de 3 años en la actualidad. Embarazo y parto normal. Rama materna: Displasia metafisaria. Exploración física y desarrollo ponderoestatural normal hasta el año de vida. Tras el inicio de la marcha comienza a detectarse disminución en la velocidad de crecimiento, alcanzando a los 2 años una talla 1,2 DE, arqueamiento de las extremidades inferiores en varo y relación SS/SI de 1,66, con fenotipo facial normal. En el mapa óseo se evidencia engrosamiento de la cortical y ensanchamiento metafisario de los huesos largos de las extremidades inferiores. A la edad de 3 años la talla era de 2 DE con exploración física y radiológica similar. En el estudio analítico destaca Ca: 9,9 mg/dl (n); Pi: 2,6 mg/dl (↓); FAO. 119 U/l (↑); PTH: 81,2 pg/ml (↑); 1,25 (OH)2 D: 21 μg/ml (n); Ca/Cr: 0,22 (↑), Pi/Cr. 1,52 (↑); RTP: 47,4 % (↓ ↓). Estos datos analíticos, junto con las manifestaciones radiológicas y los antecedentes familiares, llevó a realizar estudios genéticos en los que se detectó una mutación heterozigota tipo cambio de sentido en el exón 19 del gen PHEX (Xp 22.1), que aparece también en los demás familiares afectados. Esta mutación ocasiona por mecanismos no del todo conocidos, una reabsorción deficitaria de fosfatos en el túbulo contorneado proximal y una disminución en la producción de calcitriol, por lo que es responsable del raquitismo hipofosfatémico familiar ligado al X; hasta recientemente, que no había posibilidad de realizar estos estudios genéticos, no fue posible diferenciarla de la displasia metafisaria tipo Jansen. El estudio del eje somatotrópico evidenció un déficit en la secreción de GH: IGF-1: 80 ng/ml, IGFBP3: 4,2 μg/ml, GH: pico máximo de 5,53 ng/ml (tras estímulos de insulina y ejercicio + propranolol). El tratamiento médico de esta entidad consiste en el aporte de fosfatos y calcitriol, que se verá intensificado por la acción de la hormona de crecimiento.
Estudio y cuantificación de la expresión de genes placentarios implicados en RCIU
E. Barrio Ollero, M.T. Calvo, A. Romo, J. Naval y A. Ferrández-Longas
Genética Molecular. HMI Miguel Servet. Zaragoza. España.
Introducción: En situaciones de RCIU (Crecimiento Intrauterino Restringido) hemos demostrado un aumento significativo de la apoptosis en las placentas RCIU frente a las placentas normales. Este hecho ha motivado el estudio de la expresión de genes implicados en las vías apoptóticas y la cuantificación de genes que están implicados en el crecimiento y desarrollo. Los genes candidatos han sido Bcl-2, telomerasa y caspasa 3 (genes implicados en la apoptosis), CRH (estrés) y pGH (hormona de crecimiento placentaria).
Materiales y métodos: Para todos los genes diana se utilizó una PCR cuantitativa a tiempo real (LigthCycler 2.0, Roche Diagnostics S.L.). Se hizo una extracción de ARN de tejido placentario (RNAwiz, Ambion), posterior retrotranscripción a ADNc (RetroScript, Ambion) y una amplificación y cuantificación de la expresión de los genes. El análisis estadístico fue por ANOVA y se consideró significativo si p < 0,05.
Resultados: Como hallazgos más significativos hemos encontrado una expresión disminuida en el caso de Bcl-2 y CRH y un aumento en el caso de la caspasa 3, estadísticamente significativo.
Gigantismo cerebral (síndrome de Weaver)
G. González Lorenzo, J.M. Díaz Grande, R. Santolaya Machetti y L. Puig Pérez-Quevedo
Servicio de Pediatría y Radiología. Hospital de Pontevedra. Centro A.P. Baltar. Sanxenxo. Pontevedra. España.
Introducción: Paciente varón de 10 años, con diagnóstico de síndrome de Weaver (inseminación artificial, parto gemelar). El paciente presenta hipercrecimiento prenatal y posnatal, avance asincrónico de la edad ósea, facies peculiar (macrocefalia, hipertelorismo, micrognatia y orejas grandes), ventrículos cerebrales asimétricos, babeo continuo, problemas en la coordinación motora, con marcha en la punta de los pies, mala adaptación social y camptodactilia.
Antecedentes clínicos:Historia personal. Parto gemelar. Peso al nacimiento. 3.800 g. Longitud. 51 cm. Perímetro craneal: 38 cm. Historia familiar. Inseminación artificial. Longitud del hermano al nacimiento: 49 cm. Consulta inicial a los 4 años de edad, presentaba crecimiento excesivo, babeo continuo, problemas en la coordinación motora con marcha en la punta de los pies y mala adaptación social.
Examen físico: Facies característica con macrocefalia (P.C. 52,5 cm), cara redonda, occipucio plano y camptodactilia, marcha sobre la punta de los pies, hipertonía en extremidades inferiores, pobre pero lenguaje normal, talla: 111 cm (> P97). Volumen testicular: 2 ml.
Estudio endocrinológico: TSH, FT4, GH, IGF-I, IGFB-P3 en rangos normales.
Estudio molecular: Descartado cromosoma X frágil. Gen NSD1. pendiente de estudio. Tanto el síndrome de Weaver como el de Sotos tienen similares características, con pequeñas diferencias. Actualmente se considera que ambos síndromes son alélicos.
Destacamos en nuestro paciente: La circunstancia de que inicialmente la edad ósea estaba retrasada con respecto a la cronológica, lo cual dificultó el diagnóstico. Más tarde se igualó a la cronológica y, actualmente, tiene la edad ósea adelantada con respecto a la cronológica (véase la evolución de la edad ósea). Otros datos que hay que reseñar son la discrepancia entre la edad ósea del carpo y la metacarpofalángica y la presencia de babeo continuo, más frecuente en el síndrome de Sotos.
RM cerebral: Asimetría en el tamaño ventricular (ligeramente mayor el ventrículo lateral derecho) probable variante de la normalidad; no se evidencian otras anormalidades patológicas.
En este momento, nuestro paciente tiene 10 años, y su talla es de 159 cm, su peso de 67.600 g, P.C. 52,5 cm. Su hermano: talla: 135 cm, peso: 27.000 g. y P.C. 52,5 cm.
Evolución: El paciente, después de rehabilitación continua ha mejorado su coordinación motora y adquirió habilidades que antes no tenía, tal como la de sostenerse en un solo pie.
Síndrome de Simpson Golabi Behmel. Una nueva familia con una mutación intrónica
L.F. Magano Casero, R. Gracia, M. Segovia, R.M. Valdez, I. Incera, P. Arias y P. Lapunzina
Genética Molecular. Bioquímica. Endocrinología. Hospital Universitario La Paz. Madrid. España. Instituto Nacional de Genética. Buenos Aires. Argentina.
El síndrome de Simpson-Golabi-Behmel (SGBS) es un síndrome ligado al cromosoma X caracterizado por un sobrecrecimiento prenatal y posnatal (gigantismo) que incluye macroglosia, macrosomía, anormalidades renales y esqueléticas y un riesgo elevado de tumores embrionarios, como el de Wilms y el de hígado. Las deleciones y/o mutaciones que implican el gen glipican-3 (GPC3) en Xq26 están asociadas al síndrome (SGBS). GPC3 codifica una proteína de la familia de los proteoglucanos, glipican-3, que se supone que desempeña un papel importante en control del crecimiento en los tejidos mesodérmicos embrionarios, en los cuales se expresa selectivamente. Parece formar un complejo con el IGF-2, y puede modular la acción de éste. Nosotros estudiamos el gen GPC3 en una familia con SGBS. El caso índice y su tío materno presentan una mutación intrónica (IVS2 + 1 G > A) que predecimos que produce una pérdida de función de la proteína glipican-3.
Síndrome de Turner. Respuesta al tratamiento con hormona del crecimiento
T. Arévalo Saade, P. Cabanas, L. Castro Feijoó, M. Fuster, J. Barreiro Conde y M. Pombo
Endocrinología Pediátrica y Crecimiento. Hospital Clínico Universitario y Universidad de Santiago de Compostela. España.
Introducción: El síndrome de Turner (ST) es una cromosomopatía caracterizada por la ausencia total o parcial de un cromosoma sexual. La talla baja está presente en el 95 % de los casos y constituye una de las indicaciones para el tratamiento con hormona del crecimiento.
Objetivos: 1. Analizar la variación del crecimiento en las pacientes con ST en tratamiento con GH. 2. Estudiar la interacción entre factores genéticos, cariotipo, anomalías congénitas y el crecimiento de estas pacientes. 3. Verificar las ventajas del uso de hormona de crecimiento en el ST.
Material y métodos: Estudio retrospectivo realizado desde enero de 1984 hasta diciembre de 2004, de pacientes con ST, en tratamiento con hormona de crecimiento (do: 0,035 mg/kg/ día) y que alcanzaron talla final. Se analizaron los siguientes aspectos: a) demográficos; b) cariotipo; c) talla diana; d) evolución del crecimiento (SDE). Se utilizó el programa SPSS 11.0 para el análisis estadístico.
Resultados: Un total de 22 pacientes han alcanzado su talla final. Todas ellas cumplieron tratamiento con hormona de crecimiento durante una media de 7 años, y se observó una marcada reducción del SDE en los primeros 2 años. La variación SDE de talla al final del tratamiento fue 0,5. El 22 % alcanzaron tallas mayores de 150 cm. En la tabla comparamos las características auxológicas y genéticas entre el grupo (n = 5) que alcanzó una talla adulta igual o superior a 150 cm (grupo A) y las que alcanzaron tallas inferiores a 150 cm (n = 17) (grupo B).
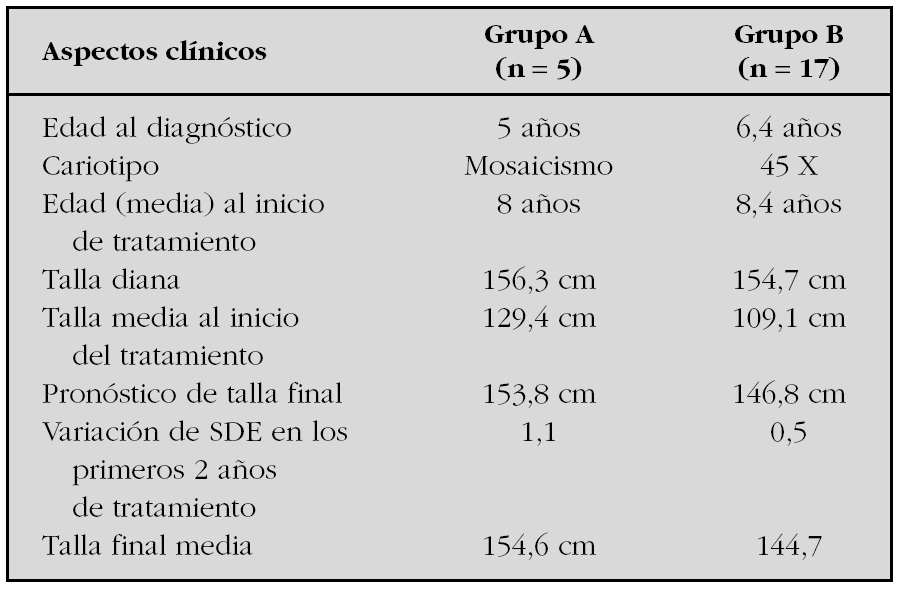
Al analizar estos datos observamos que la talla diana y la edad de inicio de tratamiento con GH no muestran diferencias significativas entre ambos grupos. No obstante, en el grupo A, el SDE de talla antes del inicio del tratamiento así como la respuesta a la terapia con GH ha sido mejor que en el grupo B, lo cual podría estar relacionado con las características del cariotipo. Las pacientes 45X muestran tallas inferiores antes y después del tratamiento.
Conclusiones: En nuestra serie, la variación de la respuesta al tratamiento con GH de las pacientes con síndrome de Turner parece estar influenciada por características genéticas (cariotipo).
DIABETES
Insulina glargina y control metabólico de la diabetes infantil
M.D. Esteban Oliva, P. Sol Ventura, J. Bel Comos y C.R. Gonzalo de Liria
Servicio de Pediatría. Hospital Universitari Germans Trias i Pujol. Badalona. Barcelona. España.
Introducción y objetivos: La glargina (lantus) es un análogo de la insulina de semivida larga recientemente aprobado para el tratamiento de la diabetes y de uso creciente en la población diabética infantil dada su capacidad teórica de mejorar el control metabólico, aunque aún existe poca evidencia práctica. Presentamos aquí la experiencia con glargina en nuestro centro en el último año en el subgrupo de pacientes de peor control metabólico con pauta clásica.
Métodos: Trabajo observacional retrospectivo con 19 pacientes de edades comprendidas entre 2 y 18 años (edad media 13,73 ± 3,81; 10 niñas y 9 niños) a los que durante el último año se ha cambiado la insulina NPH por glargina por mala evolución. Se han determinado los requerimientos absolutos de insulina y en relación al peso, la hemoglobina glicada (HbA1c) y el índice de masa corporal (IMC) basalmente y a los 3, 6 y 9 meses. Se analizan los datos mediante t de Student con programa estadístico SPSS (versión 12.0).
Resultados: Antes del cambio, los requerimientos de insulina con relación al peso eran de 1,12 ± 0,29 U/kg/día (pauta clásica) mientras que con glargina han sido de 0,99 ± 0,23 (al inicio), 0,96 ± 0,25 (a los 3 meses), 0,98 ± 0,28 (a los 6 meses) y 0,94 ± 0,25 (a los 9 meses). El porcentaje de insulina rápida y lenta era inicialmente de 31 % rápida ± 0,14 y 68,8 % ± 10,2 de NPH y al cambiar a lantus fue de 39 % ± 7,8 de rápida y 60,9 % ± 7,74 de glargina al inicio, 35,2 % ± 10 de rápida y 64,64 % ± 10 lantus (3 meses), 36,3 % ± 10 y 62,8 % ± 10 (6 meses) y 38 % ± 12 y 61 % ± 12 (9 meses). La HbA1c por su parte pasó de 8,86 ± 1,23 g/l (en el momento previo de paso a glargina) a 8,14 ± 1,15 (a los 3 meses), 8,13 ± 1,3 (a los 6 meses) y 8,57 ± 1,36 g/l (a los 9 meses). El IMC inicial fue de 22,5 ± 2,89, a los 3 meses y a los 6 meses 22,6 ± 2,8 y 21,47 kg/m 2 a los 9 meses. Ningún paciente presentó complicaciones graves ni hipoglucemias importantes.
Conclusiones: Los valores de HbA1c disminuyeron de manera estadísticamente significativa a los 3 y 6 meses del cambio (p 0,17 y 0,06, respectivamente) mientras que la reducción no fue estadísticamente significativa a los 9 meses. El IMC, por su parte, tiende a disminuir durante el seguimiento aunque no alcanza significación estadística. Estos datos sugieren que la glargina es eficaz para la optimización del control metabólico de la diabetes infantil sin complicaciones importantes, aunque hay una tendencia hacia la pérdida de este efecto beneficioso inicial a lo largo del seguimiento. Son necesarias series clínicas más amplias con períodos de seguimiento prolongados para aclarar estos resultados.
Insulina glargina en niños y adolescentes con diabetes tipo 1 en tratamiento intensivo
E. García García, R. Sánchez-Pérez, P. Cortés-Mora, E. López-Ruzafa, P. Aguilera-Sánchez y M. Rodríguez-Martínez
Servicio de Endocrinología Pediátrica.Complejo Hospitalario Torrecárdenas. Almería. España.
Objetivos: 1. Evaluar el tratamiento con una pauta insulínica con glargina en niños y adolescentes. 2. Valorar el grado de satisfacción de los pacientes y sus padres con esta nueva pauta.
Pacientes y métodos: Presentamos 42 pacientes con diabetes tipo 1, 27 mujeres, edad media al inicio 6,8 años (rango: 1,2-13,2), edad media al inicio del tratamiento con glargina 12,8 años (rango: 7,0-17,7), tiempo de evolución medio 6,1 años (rango: 2,0-11,9). Las indicaciones de la nueva pauta fueron mal control metabólico, hipoglucemias frecuentes y sobrepeso con la pauta intensiva con 2 o 3 dosis de NPH, que fueron sustituidas por una dosis de glargina. La satisfacción de pacientes y padres con el tratamiento de la diabetes se valoró con la escala publicada por Boot et al. Prueba estadística t de Student, nivel de significación p < 0,05.
Resultados: Las variables se expresan como media (desviación estándar). Tras un período medio de 7,3 meses (rango: 6-9 meses), la nueva pauta disminuyó la concentración de HbA1c (7,62 % [0,86] frente a 8,11 % [0,83], p < 0,001), la dosis de insulina (0,99 [0,24] U/kg/día frente a 1,07 [0,23]; p < 0,001) y el SDE (score de desviación estándar) del índice de masa corporal (+0,57 [0,93] frente a +0,72 [1,07]; p = 0,001), mientras que aumentó el grado de satisfacción con el tratamiento de los niños (+44,5 [18,8] puntos en la escala frente a 9,9 [26,8]; p < 0,001) y de sus padres (+42,0 [17,9] puntos en la escala frente a 20,8 [29,1]; p < 0,001).
Conclusiones: 1. La pauta insulínica con glargina mejora el control metabólico y disminuye el peso y la dosis de insulina en niños y adolescentes en tratamiento intensivo. 2. También mejora el grado de satisfacción de pacientes y padres con el tratamiento de la diabetes.
Intervención psicoeducativa con niños y adolescentes con diabetes tipo 1
E. Mora Gómez-Cacerrada, P. Giralt Muiña, M. Beléndez Vázquez, M.J. Ballester Herrera, L. Zapata García y J. Giralt Contreras
Fundación de Castilla-La Mancha para la Diabetes. Ciudad Real. Hospital de Alicante. Universidad de Alicante. España.
Introducción: Las intervenciones dirigidas al control de la diabetes tipo 1 que utilizan métodos didácticos junto con estrategias de modificación conductual han demostrado conseguir notables mejoras en la adherencia al tratamiento y calidad de vida de los pacientes. En el presente trabajo presentamos una intervención que combina modelos de enseñanza-aprendizaje basados en el constructivismo con el entrenamiento en estrategias de afrontamiento, en autocontrol y el empleo del contrato conductual.
Objetivos: Se desarrolló un programa de educación mediante el cual se pretendía: 1. Que los niños y adolescentes fueran sujetos activos de su salud, protagonistas del proceso y progreso de la enfermedad y su tratamiento. 2. Mejorar la calidad de vida del paciente. 3. Conseguir un buen control metabólico, y que éste fuera sostenido. 4. Aumentar la adhesión al tratamiento y la normalización de los hábitos que esto conlleva. 5. Mejorar el conocimiento de los niños y adolescentes sobre el tratamiento de la diabetes.
Pacientes y método: Participaron 25 pacientes con diabetes tipo 1 (10 niños de 8 a 12 años, y 15 adolescentes de 13 a 15). Para ambos grupos de edad se aplicaron intervenciones similares en cuanto a su estructura y duración (siete sesiones de 90 min) pero diferentes en metodología didáctica y contenidos (entre otras técnicas se utilizó el juego didáctico para los niños y el entrenamiento en afrontamiento con adolescentes). Se aplicaron los siguientes instrumentos de evaluación: cuestionarios de conocimientos (Beléndez, Ros y Bermejo), cuestionarios para medir la calidad de vida: PedsQL (Varni), en sus versiones 3.0 específico para diabetes y 4.0 sobre salud general, cuestionario de responsabilidad en las tareas. Propio. Basado en el trabajo y cuestionario de Barbara J. Anderson et al (1990), cuestionario de adherencia al tratamiento Autocuidado de la diabetes, propio, hoja de datos sociodemográficos y clínicos. Anotaciones de campo. Evaluación observacional.
Resultados: Al término de la intervención, y comparados los datos obtenidos al comienzo y al final de la misma, tanto niños como adolescentes mostraron una mejora, en algunos casos significativa, de las variables estudiadas. Calidad de vida (niños p = 0,01, adolescentes p = 0,6); conocimientos (niños p = 0,55, adolescentes p = 0,2); responsabilidad (niños p = 0,00, adolescentes p = 0,11); y adherencia al tratamiento (niños p = 0,1, adolescentes p = 0,69).
Conclusiones: Una intervención terapéutica que combine métodos didácticos con estrategias de modificación de conducta obtiene como resultados a corto plazo (2 meses) una mejora significativa en la calidad de vida y el grado de responsabilidad de los niños, y un progreso en el nivel de conocimientos y la adherencia al tratamiento de niños y adolescentes.
Medición de la cetonemia en diabetes tipo 1 en una colonia de verano
J.P. López Siguero, J.A. Moreno, M.V. Borrás, J.L. Pinzón, J.I. Brea, A. del Pino, M. García y M. Parramón
Hospital Materno-Infantil de Málaga. Hospital de Granollers. Barcelona. Hospital Clínico de Málaga. Abbott Científica. España.
Introducción: La utilidad de la medida de cuerpos cetónicos en la infancia y adolescencia ha sido descrita en numerosos trabajos. En un ambiente controlado, como es una colonia de verano, por un lado los horarios de comidas son muy regulares, pero por otro lado, los niños desarrollan una gran actividad física. Por ello, es conveniente hacer un seguimiento no sólo de los perfiles de glucemia, sino también de los cuerpos cetónicos en ciertas situaciones con un dispositivo que permita obtener dichos valores en sangre capilar y con un pequeño volumen de gota.
Objetivos: 1. Valorar la repercusión de una actividad física moderada de 45 min sobre cetonemia y la cetonuria, así como sobre la glucemia. 2. Valorar la relación entre glucemias, cetonemias y cetonuria durante la colonia en tres aspectos: a) los valores las preprandiales durante 3 días de seguimiento; b) aquellos desayunos con cetonemia por encima de 0,5 mmol/l y su evolución a la hora y a las 3 h, y c) cetonemias y cetonurias de las glucemias preprandiales altas (> 250 mg).
Pacientes y métodos: Se analizaron los datos de 47 participantes (25 varones y 22 mujeres) de una colonia de verano de 14 días. La edad media era de 11,7 ± 1,8 años, con un IMC de 18,3 ± 3,9 y 3,0 ± 2,7 años de evolución de la diabetes. La dosis media de insulina era de 0,8 ± 0,4 U/kg/día, el número de dosis de 2,7 ± 0,7, y la HbA1c media de 7,7 ± 1,5 %. Para la medida de las glucemias y cetonemias se utilizó el sistema Optium® (Abbott Diabetes Care) y para las cetonurias se usaron las tiras Ketodiastix® (Bayer). Para el análisis estadístico se usó el programa SPSS 11,5. Se hizo un análisis descriptivo de las variables, expresándolas con su media, desviación estándar, y rango. El punto de corte para considerar clínicamente significativo el dato de cetonemia fue de igual o superior a 0,5 mmol/l.
Resultados: 1. Los datos de cetonemia y cetonuria antes y 45 min después del ejercicio indicaron ausencia de elevaciones significativas de cuerpos cetónicos. 2. Valores preprandiales durante 3 días de seguimiento: El 17,02 % de los participantes tuvo niveles de cuerpos cetónicos iguales o superiores a 0,5 mmol/l. La media de todas las glucemias fue 156,87 ± 87,8 mg/dl y de aquellas glucemias que cursaron con cetosis igual o superior a 0,5 mmol/l fue de 174,5 ± 64,7 mg/dl. Se encontraron más cuerpos cetónicos en la mañana que durante el resto del día (0,12 ± 0,17 mmol/l frente a 0,02 ± 0,05 mmol/l; p < 0,001) y todas las cetonemias igual o superior a 0,5 mmol/l ocurrieron en el desayuno. Todas las cetonemias se resolvieron en la primera hora, excepto una que se resolvió en 3 h. Se registraron 29 glucemias preprandiales altas (> 250 mg/dl) con más frecuencia (59 %) en las cenas, de las que el 10 % tuvieron valores de cetonemia iguales o superiores a 0,5 mmol/l. Hubo un porcentaje elevado de pacientes en los que no se pudo conseguir la muestra para determinar la cetonuria: 24,5 % en el seguimiento a 3 días, 28,4 % en los datos de desayuno con cetonemia y 58,6 % en los resultados de preprandiales altas.
Conclusiones: 1. La ausencia de elevaciones significativas de cuerpos cetónicos tras 45 min de ejercicio nos lleva a la siguiente recomendación clínica: si no existe cetonemia, un ejercicio moderado de 45 min no la produce. 2. Todas las cetonemias se produjeron en el desayuno, con glucemias elevadas, y desaparecieron rápidamente. 3. Un porcentaje importante de datos de cuerpos cetónicos en orina no se pudieron determinar por falta de muestra. Esta dificultad, que es particularmente importante en cetosis más avanzadas en las que hay un cierto grado de deshidratación, se salva por la posibilidad de medir cuerpos cetónicos en sangre.
Utilización de monitorización continua de glucosa en diabetes tipo MODY
R. Cebrián Rubio, M. Gussinyer, M. Clemente, M. Losada, E. Armengol, M. Albisu, D. Yeste y A. Carrascosa
Unidad de Endocrinología Pediátrica. Hospital Universitario Vall d'Hebron. Barcelona. España.
Objetivo: Conocer el perfil glucémico de pacientes afectados de diabetes tipo MODY.
Material y métodos: Se estudiaron 5 familias afectadas de diabetes tipo MODY. En dos de ellas no se encontró mutación genética aunque cumplían criterios diagnósticos de MODY, las otras 3 familias presentaron mutación en el gen de la glucocinasa 7p15-p13: exón 8, exón 10 e intrón 6. En cuatro de estas familias, en total 9 sujetos con edades comprendidas entre los 3 y 47 años, se realizó durante 3 días monitorización continua de glucosa mediante el Continuous Glucose Monitoring System (CGMS). Durante la monitorización algunos pacientes seguían una dieta restrictiva en hidratos de carbono de absorción rápida y otros, dieta libre.
Resultados: Todos presentan glucemias en ayunas superiores a 100 mg/dl. Todos los pacientes presentan glucemias superiores a 150 mg/dl en algunas ocasiones. No necesariamente en período posprandial.
Conclusiones: Parece que el perfil glucémico no varía con la edad. Hay una diferencia no significativa en el perfil glucémico siguiendo o no una dieta restrictiva en hidratos de carbono de absorción rápida.
Monitorización continua de glucosa en niños con infusor de insulina
E. García García, R. Sánchez-Pérez, J. Ramos-Lao, E. López Ruzafa, P. Aguilera-Sánchez y M. Rodríguez-Martínez
Endocrinología Pediátrica. Complejo Hospitalario Torrecárdenas. Almería. España.
Introducción: El tratamiento con infusor de insulina mejora el control glucémico y disminuye el riesgo de hipoglucemia en diabetes tipo 1.
Objetivo: Valorar la concentración de glucemia y la presencia, duración y sintomatología de las hipoglucemias e hiperglucemias con el uso de un monitor continuo de glucosa (CGMS de Minimed) en niños y adolescentes con diabetes tipo 1 tratados con infusión de insulina, así como la tolerancia de esta prueba en estos pacientes.
Pacientes y métodos: Se estudian 8 pacientes (4 varones) con diabetes tipo 1 tratados con infusor de insulina, edad media 10,6 años (rango: 3,2-14,2), tiempo de evolución de la diabetes medio 6,3 años (rango: 2,1-11,5), tiempo medio de tratamiento con bomba 12,0 meses (rango: 8,0-18,0), dosis de insulina media 0,85 U/kg/día (rango: 0,55-1,01) y último nivel de HbA1c medio 7,1 (rango: 6,4-8,5).
Resultados: Los variables se expresan como media (desviación típica). El número de horas registradas fue de 52,5 (27,8), con un número de lecturas del sensor de 599 (344). La concentración de glucosa intersticial captado por el sensor fue 196,3 mg/dl (55,4). El tiempo en hipoglucemia (< 70 mg/dl) fue el 5,3 % (7,5) del total y el tiempo en hiperglucemia (> 180 mg/dl) fue el 51,5 % (24,9) del total. En ningún caso se detectaron hipoglucemias subclínicas. El sensor no fue tolerado por el niño preescolar de 3 años.
Conclusión: Los niños y adolescentes con infusor de insulina muestran un perfil glucémico muy irregular y sólo una pequeña parte del tiempo consiguen estar en normoglucemia. Nuestros pacientes conservan la sintomatología de alarma frente a la hipoglucemia y en general toleran bien el CGMS.
Factores que influyen en el control metabólico de los niños y adolescentes con diabetes
F. Manzano Recio, J. Prieto Veiga, J. Cedeño Montaño y M. Martín
Servicio de Pediatría. Endocrinología Pediátrica. Hospital Clínico de Salamanca. España.
Introducción: La organización e incorporación de niños y adolescentes con diabetes en un registro permite investigar la cantidad y calidad de los cuidados recibidos. En nuestro hospital tenemos un contacto estrecho con las familias de los niños con diabetes; mantienen una consulta formal cada 3 meses, junto con un rápido sistema de ayuda ante cualquier problema que surja: reajuste del régimen de insulina, alimentación, etc.
Objetivos: El objetivo fundamental de este trabajo es determinar el control metabólico de los niños menores de 15 años de edad con diabetes mellitus tipo 1, y los factores y circunstancias personales, familiares y sociales que pueden afectar a su control metabólico.
Pacientes y métodos: Los niños con diabetes acuden a revisión a la consulta de endocrinología infantil de manera sistemática cada 3-4 meses. Previamente y ese mismo día, en todos, se realiza la HbA1c mediante el método DCA 2000. El presente trabajo es un estudio prospectivo en el que se analizan las HbA1c de los últimos 4 años (2001-2004) en menores de 15 años, y se comparan con circunstancias personales o familiares en las que viven, con el fin de determinar si influyen o no en su control metabólico.
Resultados: Nuestro registro actual (a diciembre de 2004), cuenta con 51 niños con diabetes menores de 15 años. La duración media de la diabetes es de 4,21 años. La tabla siguiente muestra diferentes parámetros que relacionamos con la HbA1c media que los niños obtienen en cada revisión.

Conclusiones: 1. Los factores más relevantes encontrados que parecen influir en un peor control metabólico son: la edad de la madre en el embarazo, el grupo de niños de menor edad, las niñas en general y la etapa puberal de los pacientes. 2. Aunque globalmente la HbA1c media en nuestros niños es aceptable (7,88 %), siempre se puede mejorar, por lo que necesitamos estrategias para optimizar recursos como pieza clave que conduzca a alcanzar el objetivo de HbA1c media igual o inferior a 7,5 %.
Calidad de vida en adolescentes con diabetes mellitus tipo 1
I. Riaño Galán, M. Oyarzábal Irigoyen, A. Calvo Gómez-Rodulfo, M. Chueca Guindulain, S. Berrade Zubiri y F. Rivas Crespo
Servicio de Endocrinología Pediátrica. Hospitales Universitario Central de Asturias. Oviedo. Virgen del Camino. Pamplona. España.
Introducción: La diabetes tipo 1 (DM1), como enfermedad crónica, interactúa continuamente con el paciente, su familia y entorno social. La calidad de vida relacionada con la salud (CVRS) es un concepto multidimensional que incluye aspectos físicos, psicológicos y sociales que recoge la percepción subjetiva e individual de la propia salud.
Objetivo: Evaluar el impacto de la DM1 en la calidad de vida de adolescentes de nuestro medio, percibida por ellos mismos y por sus padres y profesionales sanitarios que los han asistido.
Pacientes y métodos: Se estudiaron 102 adolescentes con DM1 (51 varones) de dos centros hospitalarios especializados en su asistencia, con edad media de 15,5 años (11-20 años), tiempo de evolución 6,7 años (1-17 años) y cuya glucohemoglobina media era del 7,76 % (5-11,9 %). Se les aplicó el cuestionario Hvidore, específico para niños y adolescentes con diabetes (validado en español), que consta de tres tests. Uno para el paciente, con 52 ítems subdivididos en cuatro dimensiones: impacto de la enfermedad, preocupaciones acerca de la diabetes, satisfacción con la vida y percepción general de salud. Otro test para padres (5 ítems) mide la carga que les supone la diabetes de sus hijos. El tercero evalúa la percepción que tienen los profesionales sanitarios dedicados a asistirlo. Se compararon los resultados en relación al sexo, edad, tiempo de evolución y control metabólico.
Resultados: Impacto de la DM1: El 84,3 % nunca o raramente se sienten enfermos físicamente. Refieren que la diabetes nunca o raramente interfiere en sus relaciones sociales y con sus amigos (91,2 %) ni en su vida familiar (73,5 %). El 28,4 % siente que sus padres son demasiado protectores. Los menores de 15 años manifiestan mejor tolerancia al manejo de la diabetes en público que los mayores (51,3 % frente al 23 %; p = 0,014). El 21,6 % se sienten limitados a menudo o siempre por la dieta. Preocupaciones acerca de la diabetes: 40,2 % refieren que nunca o raramente se sienten preocupados por las complicaciones posibles de su enfermedad. Grado de satisfacción con la vida: El 17,6 % se sienten bastante o muy insatisfechos por la carga que supone la diabetes para su familia. El 88,2 % se consideran bastante satisfechos con los conocimientos acerca de su diabetes. El 82,4 % se sienten muy o bastante satisfechos con su tratamiento habitual. La satisfacción con el tratamiento es mayor en aquellos en los que la diabetes empezó después de los 7 años frente a los diagnosticados más tempranamente (p = 0,046). El 69,6 % se encuentran muy satisfechos con la flexibilidad que tienen con la dieta. Percepción general de salud: El 81,4 % califican su salud como excelente o buena. El 91,2 % se sienten bastante o muy satisfechos con su vida en general. Cuestionario para padres: La mayoría manifiesta que sus hijos no requieren cuidados extra por sus problemas físicos o psicológicos (p = 0,001). El 70,6 % de padres expresan que el estado de salud de sus hijos es bueno o muy bueno y consideran que su calidad de vida es muy buena o buena. Uno de cada dos padres experimenta intensa o gran preocupación por la salud de su hijo a largo plazo. Cuestionario para profesionales: Los profesionales sanitarios sólo califican de grande o intensa la carga de los padres por la salud de sus hijos en el 21,5 %. Además, estiman mejor cumplimiento del tratamiento en aquéllos con menor tiempo de evolución (p = 0,048) y con mejor control metabólico (p = 0,005). La valoración de la calidad de vida de los pacientes realizada por los padres y por los profesionales sanitarios es similar. El control metabólico es mejor en los de menor tiempo de evolución (p = 0,016). El 88,9 % de los que tienen mejores cifras de glucohemoglobina (< 7 %) refieren que la diabetes nunca interfiere sus relaciones sociales y con sus amigos frente al 64,1 % de los que tienen peor control (p = 0,034). No se encuentran diferencias entre chicos y chicas.
Conclusiones: 1. Los adolescentes con DM1 tienen una CVRS excelente o buena. 2. Estiman que la enfermedad afecta escasamente a su vida diaria y a su relación social y familiar. 3. Perciben que su salud es excelente o buena, aunque esta percepción se atenúa con el tiempo de evolución. 4. Un alto porcentaje de adolescentes no se preocupan, o lo hacen raramente, de las complicaciones posibles de la enfermedad.
Cetoacidosis diabética: evolución de la potasemia
E. Heras Martín, M. Sánchez, J.M. Mengíbar, A. Trujillo, G. Hollenberg, J. Macià y F. Castillo
Servicio de Pediatría. Hospital Universitario Josep Trueta. Girona. España.
Introducción: La cetoacidosis diabética (CAD) determina depleción variable de K corporal, que en estudios efectuados en adultos es típicamente del 20 %. El tratamiento de la CAD con rehidratación e insulina origina un rápido descenso en el valor de la potasemia plasmática, particularmente durante las primeras horas de tratamiento.
Objetivo: Bajo una pauta estándar de fluidos (3,5-4 l/m 2/día), de aporte de K inicial (40 mEq/l en la solución de rehidratación) y de aporte de Na (75 mEq/l), analizar que factores se hallaron asociados a las variaciones del K entre las 6 y 18 h de evolución de los inicio pediátricos ingresados en nuestro hospital.
Pacientes y métodos: Estudio retrospectivo basado en la revisión de historias clínicas. Población en estudio: todos los inicios diabéticos menores de 14 años ingresados en nuestra UCI-P en situación de CAD desde enero de 1999 hasta diciembre de 2002. Variables en estudio: gradiente de K (K al ingreso menos el K más bajo en las primeras 6-18 h de ingreso), gradiente de Na: mismo procedimiento, pH al ingreso, glucemia al ingreso, número de horas en que se utilizó SSF como solución única, número de horas de bomba de insulina, horas hasta la corrección del pH y creatinina al ingreso. Métodos estadísticos: se aplicó la t de Student para medidas repetidas y la prueba no paramétrica de Wilcoxon, así como pruebas de correlación simple (Pearson y la prueba no paramétrica de Spearman) y de regresión lineal múltiple. Los resultados se expresan en medias ± 2 EE (IC 95 %). Se ha considerado significativa una p < 0,05. Fluidoterapia: déficit calculado por reponer en 48 h. Aporte de K (una vez establecida diuresis): si potasemia 3,5-5,5 mEq/l: 40 mEq/l en la solución de rehidratación, si potasemia < 3,5: 60 mEq/l. Insulinoterapia 0,1 U/kg/h hasta la resolución de la acidosis.
Resultados: Un total de 14 niños iniciaron en situación de CAD, con una edad entre 1 y 13 años (x: 8,7 ± 3,7 años). La potasemia obtenida entre las 6 y 18 h de ingreso (3,43 ± 0,40 mEq/l) fue significativamente más baja (p = 0,002) que la del diagnóstico (4,29 ± 0,33 mEq/l). Aplicando técnicas de regresión lineal múltiple y ajustando las variables entre sí, se crea un modelo que permite correlacionar el gradiente de potasio obtenido entre el ingreso y las primeras 6-18 h de tratamiento, de forma positiva con el número de horas en que se aplicó la bomba de insulina (coeficiente β estandarizado 0,694; p 0,005) y con la glucemia al diagnóstico (β 0,455; p 0,01); de forma significativamente negativa con el número de horas hasta la corrección del pH (β 0,486; p 0,025) y con el valor de creatinina al ingreso (β 0,532; p 0,012).
Discusión: En nuestra serie el gradiente de potasio obtenido entre las 6 y 18 h de ingreso se correlacionó de forma positiva con el número de horas en que se aplicó la bomba de insulina y con la rapidez de corrección de la acidosis. Un mayor grado de deshidratación y una menor tasa de filtración glomerular consecuente, reflejados por un mayor valor de creatinina plasmática, pueden ser un exponente de una menor eliminación de cetoácidos y potasio urinarios, con una corrección más lenta de la acidosis. Si bien el rápido descenso en las primeras horas de tratamiento es debido fundamentalmente a la reentrada "insulina-mediada del potasio dentro del compartimento intracelular; creemos importante destacar estos resultados, que permitirían prever un descenso rápido del valor de la potasemia plasmática en aquellos casos que al ingreso estuviesen menos deshidratados con una creatinina plasmática normal o poco aumentada y, por tanto, también con una corrección más rápida de la acidosis.
Cetoacidosis en el diagnóstico de la diabetes. Cataluña 1989-2002
V. Borrás Pérez, E. Sánchez, N. de Lara y C. Castell
Servicio de Pediatría, Hospital General de Granollers. Agencia de Evaluación de Tecnología e Investigación Médica. Dirección General de Salud Pública. Consejo Asesor sobre la Diabetes. Barcelona. España.
Introducción: Se ha descrito una gran variación geográfica en la frecuencia de cetoacidosis (CA) en el diagnóstico de la diabetes mellitus tipo 1 (DM1) inversamente relacionada con la incidencia de la DM1 de la población. A pesar de las mejoras de atención al paciente con DM1 la frecuencia de CA no ha disminuido en los últimos años constituyendo la mayor causa de morbilidad y mortalidad en el paciente pediátrico con DM1.
Objetivos: Cuantificar la frecuencia de la CA al diagnóstico de la DM1 en menores de 15 años en Cataluña durante el período 1989-2002. Describir la distribución de la CA: por sexo, período y grupo de edad. Analizar la tendencia temporal de la CA.
Material y métodos: Se han incluido todos los menores de 15 años residentes en Cataluña diagnosticados de DM1, según los criterios de la OMS, y con presentación de CA en el diagnóstico. Se ha definido la CA según los criterios del consenso de la Lawson Wilkins Pediatric Endocrine Society (LWPES) y de la European Society for Pediatric Endocrinology (ESPE) como hiperglucemia > 11 mmol/l con pH < 7,3 y/o bicarbonato < 15 mEq/l. La información se ha obtenido del registro de DM1 de Cataluña iniciado en 1987 e incluido en el programa colaborativo EURODIAB en 1989. El grado de exhaustividad calculado a partir de las distintas fuentes es del 98 %. Se ha calculado la frecuencia de CA global (1989-2002) y para los períodos (1989-1993; 1994-1997 y 1998-2002). Estadística: test de la chi cuadrado para la tendencia lineal.
Resultados: Durante los años 1989-2002 se han registrado 1788 nuevos casos de diabetes en menores de 15 años: 251 menores de 5 años, 588 de 5 a 9 años y 949 de 10 a 14 años. De ellos, 640 (36,7 %) presentaron CA en el diagnóstico. La distribución por edad es: 50 % menores de 5 años, 36,1 % de 5 a 9 años y 33,4 % de 10 a 14 años. En la tabla adjunta se refleja el número de casos con CA y la proporción global y por períodos en los diferentes grupos de edad (tabla 1).

Conclusiones: Algo más de un tercio de los diabéticos menores de 15 años empiezan con CA, llegando al 50 % en los menores de 5 años, sin diferencias en cuanto al sexo. La frecuencia de CA entre 1989 y 2002 ha experimentado un aumento estadísticamente significativo en los menores de 15 años.
Múltiples dosis de insulina en la diabetes tipo 1: comparación de tres programas diferentes de administración
G. Carreras González, J. Torrubiano, S. Brió, A. Sánchez, C. López, L. Sojo y J. Cubells
Pediatría y Endocrinología. Hospital Sant Pau. Barcelona. España.
Introducción: La insulina regular preprandial y NPH nocturna se considera la pauta estándar de múltiples dosis. En los últimos años, tras los cambios en la comercialización de la insulina regular y la comercialización de la insulina glargina, se han desarrollado nuevas pautas de administración de insulina.
Objetivo: Comparar 3 pautas de administración de insulina en adolescentes con diabetes tipo 1.
Material y métodos: Estudiamos 16 adolescentes (9 chicos) con diabetes tipo 1, con edad media de 15 ± 2,5 años y un tiempo de evolución medio de la enfermedad de 6,1 ± 3,8 años. Todos estaban tratados con insulina regular preprandial y NPH nocturna (pauta 1). De manera ambulatoria se cambiaron a mezclas fijas de lispro y NPL preprandiales (MIX 50 predesayuno y precomida y MIX 25 precena; pauta 2) durante 12 ± 4 meses. Finalmente, se transfirieron a análogo de rápida preprandial e insulina glargina nocturna (pauta 3) durante 6 ± 2,5 meses. Se analizan el control glucémico (HbA1c), los requerimientos de insulina (U/kg/día), la distribución de la insulina durante el día y la repercusión sobre el peso. El análisis estadístico se realizó mediante los tests de Friedman y de Wilcoxon (SPSS 11,5.2 para Windows).
Resultados: Al cambiar de la pauta 1 a la 2, la HbA1c baja de 9,1 ± 1,5 % a 8,4 ± 1,1 % (p < 0,05), aumentan los requerimientos de insulina (0,87 ± 0,2 U/kg/día frente a 1,04 ± 0,2 U/kg/día; p < 0,05) y aumenta el IMC (20,0 ± 2,4 kg/m 2 frente a 21,4 ± 2,7 kg/m 2; p < 0,01). Al cambiar posteriormente a la pauta 3, se produce una nueva mejoría de la HbA1c (7,6 ± 0,9 %; p < 0,01 frente a pauta 2), pero no se modifica el IMC (21,5 ± 2,7 kg/m 2) y los requerimientos de insulina vuelven a bajar (0,94 ± 0,2 U/kg/día) a valores similares a los de la pauta 1. Considerando la insulina basal como la dosis de NPH en la pauta 1, la dosis de NPL en la pauta 2 y la dosis de glargina en la pauta 3, respecto a la dosis total, observamos que es más alta en la pauta 2 (62 ± 3 % frente a 48 ± 11 % en la pauta 1 y frente a 47 ± 8 % en la pauta 3; p < 0,001).
Conclusión: las pautas con múltiples dosis de insulina con análogos de insulina rápida e insulina glargina son más eficaces que las pautas con mezclas fijas de insulina lispro y NPL en adolescentes con diabetes tipo 1.
Niños diabéticos en el contexto de familias desestructuradas: ¿una forma de maltrato?
L. Sentchordi Montané, A. Aragonés Gallego, L. Goenechea, A. Crespo Alonso, M.D. Sánchez-Redondo y B. Martín Sacristán
Servicio de Pediatría y Psiquiatría. Hospital Virgen de la Salud. Toledo. España.
Introducción: La diabetes mellitus tipo 1 es una enfermedad crónica que requiere para su correcto tratamiento de la colaboración de muchos profesionales. Pero sin duda lo más importante es la motivación y la capacidad de autocontrol de los pacientes, basada en aspectos psicológicos y sociales, y que son los atributos más difíciles de proporcionar. En el caso de la diabetes infantil son los padres los responsables del control. La inestabilidad familiar se suele traducir en mal tratamiento metabólico, pero en ocasiones aparecen situaciones complejas, que llevan a los niños a hospitalizaciones continuas inaceptables. Presentamos 2 pacientes cuya situación familiar les lleva a múltiples descompensaciones y en quienes la intervención multidisciplinar no consigue buenos resultados.
Caso 1: Niño diagnosticado de diabetes a los 7 años. Fue dado de alta tras control metabólico y educación a la madre, con adhesión insuficiente por parte de ésta. Durante el año que sigue al inicio precisa al menos nueve ingresos por descompensación, cinco de ellos en UCIP por cetoacidosis, todos ellos derivados del mal tratamiento. Durante este tiempo se ha insistido en la educación diabetológica, se han probado diferentes pautas de tratamiento y se ha consultado con otra unidad de diabetes infantil. Así mismo, se ha solicitado valoración y tratamiento psicológico del niño y la familia. La madre, pese a todo, continúa mostrando actitudes irresponsables y conductas arriesgadas que empobrecen la calidad de vida de su hijo.
Caso 2: Niño seguido desde los 6 años por diabetes tipo 1. Durante los primeros 2 años tras el inicio presentó buen control metabólico y buena adherencia al tratamiento. A los 8 años comenzó con descompensaciones continuas que requirieron ingreso en ocho ocasiones en un período de 3 meses, incluyendo cetoacidosis graves. Tras descartar enfermedades desencadenantes, se insistió en educación diabetológica así como seguimiento estrecho en consulta externa y telefónicamente, pero no se pudo evitar continuos reingresos. Se sospechó entonces incumplimiento o manipulación deliberada del tratamiento por parte de la madre, por lo que se solicitó colaboración por parte de una unidad de diabetes infantil en otro centro, de los servicios de psiquiatría y psicología, pediatra de área, asistente social, gabinete jurídico y juzgado de guardia. Pese a los esfuerzos no se pudo demostrar la hipótesis, aunque disminuyó el número de ingresos. La situación se resolvió con el fallecimiento accidental del padre adoptivo. A partir de este momento el niño no volvió a requerir ingreso y presenta en la actualidad buen control metabólico.
Conclusiones: 1. El mal control de la diabetes en la edad pediátrica podría llegar considerarse una forma de maltrato infantil. 2. Las hemoglobinas glucosiladas altas repetidas, la omisión de controles glucemia capilar, la inasistencia a las revisiones y los ingresos repetidos deben poner a los especialistas en alerta. 3. La presencia de distocias sociales y patología psiquiátrica en la familia convierten estos problemas en situaciones de difícil solución.
Diabetes tipo 1, tiroiditis autoinmune y enfermedad celíaca: prevalencia de su asociación y forma de presentación
F.J. Herrero Espinet, A. Champer Blasco, A. Roca Comas, M. Peñas Boira, J. Sánchez Jiménez y A. Ballester Martínez
Servicio de Pediatría. Corporació de Salut del Maresme i la Selva. Calella. Barcelona. España.
Introducción: Es bien conocida la asociación de enfermedad celíaca (EC) con enfermedades endocrinológicas de origen autoinmune, como la diabetes mellitus tipo 1 (DM), tiroiditis de Hashimoto (TH) o síndrome poliglandular autoinmune.
Objetivos: Conocer la prevalencia de la sociación de EC, DM y TH en nuestro medio. Investigar la forma de presentación de estas asociaciones.
Métodos: Estudio prospectivo iniciado en noviembre de 2002. Casos: Los pacientes diagnosticados de DM, TH y/o EC. Se registran: edad, sexo, año de diagnóstico de la enfermedad de inicio y de las enfermedades asociadas, en caso de haberlas, y el período de latencia entre ellas. Determinaciones realizadas: anticuerpos anti-GAD, antiinsulina (IAA), anti-IA2, antitiroglobulina (TG), antiperoxidasa tiroidea (PO), antitransglutaminasa (TTG), antiendomisio (EMA). Estudio estadístico: métodos estadísticos descriptivos.
Resultados: Se incluyen un total de 62 casos. Enfermedad de inicio: DM: 25 casos (40 %), de ellos 16 varones, edad media 11,5 ± 3,9 años (rango: 1-14). TH 17 casos (23 %), de ellos 3 varones, edad media 10,4 ± 2,59 (rango: 6-14). EC 20 casos (32 %), de ellos 7 varones, edad media 4,6 años ± 3,4 (rango: 1-13). Patología asociada: DM y EC en 2 casos, DM y TH: 2 casos. Tiempo de latencia entre la DM y la otra enfermedad: 3 años (DE ±2,31). No se observan otras combinaciones en esta serie salvo la presencia de una paciente afectada de TH con positividad de dos anticuerpos frente al páncreas sin clínica de DM.
Conclusiones: La prevalencia de asociación entre estas tres enfermedades es del 3 %. El 10 % de los pacientes con EC presentan DM. La enfermedad inicial ha sido DM en el 100 %. La asociación de DM con TH (3 %) y de DM con EC (3 %) se presenta con la misma frecuencia. El intervalo de tiempo entre el inicio diabético y el diagnóstico de otra enfermedad asociada es de 3 años (DE ±2,31).
Diabetes mellitus tipo 1. Diagnóstico y evolución
M.J. González García y F.J. Arroyo Díez
Servicio de Pediatría. Hospital San Pedro de Alcántara. Cáceres. España.
Introducción: La diabetes mellitus tipo 1 (DM1) es la enfermedad crónica más frecuente en la infancia. Parte de su importancia se basa en la asociación a otras enfermedades autoinmunes y al desarrollo de complicaciones.
Objetivo: Conocer el modo de presentación de la DM1 en los niños cacereños, el control metabólico y la aparición de enfermedades asociadas.
Pacientes y métodos: Se han incluido en el estudio los niños con DM1 controlados en las consultas externas del hospital San Pedro de Alcántara (Cáceres) en los últimos 18 meses. En el protocolo de recogida de datos se incluía: edad, sexo, edad al diagnóstico de DM1, modo de presentación, HbA1c en el momento del diagnóstico, al año de evolución y la media del último año, enfermedades asociadas y edad del diagnóstico y aparición de complicaciones.
Resultados: Se han estudiado 67 pacientes, cuya distribución por sexos es: 58,2 % niños (39 casos) y 41,8 % niñas (28 casos). La edad media del diagnóstico de la DM1 fue de 6,9 años, con un intervalo de 20 meses a 14 años. La forma de presentación más frecuente fue la cetosis (46,2 %), seguida de la cetoacidosis (23,8 %) e hiperglucemia (22,3 %), con una media global de glucemia en el inicio de 397,8 mg/l ± 156,3. La HbA1c en el momento del diagnóstico fue 10,44 % ± 2,50, al año del diagnóstico 7,22 % ± 1,2, y la media del último año 7,92 % ± 0,96. En cuanto a las enfermedades asociadas, 16 pacientes (23,8 %) presentan patología tiroidea autoinmune, 2 pacientes (2,9 %) enfermedad celíaca, y otros 2 (2,9 %) patología tiroidea + enfermedad celíaca. El tiempo medio del diagnóstico de las enfermedades asociadas fue, 4,5 años después del debut diabético para la patología tiroidea (4 pacientes se diagnosticaron al mismo tiempo que la DM1) y 1,3 años para la enfermedad celíaca. El 23,8 % de los pacientes (16 casos) han presentado alguna hipoglucemia grave en la evolución de su enfermedad y el 8,9 % (6 casos) presentan microalbuminuria incipiente; ninguno de los pacientes presenta alteración en el fondo de ojo.
Conclusiones: La HbA1c es el mejor marcador del control metabólico en la DM1, en nuestro grupo de pacientes, las cifras están por encima de los niveles óptimos para evitar las complicaciones que se asocian a esta enfermedad. La patología tiroidea es la enfermedad que se asocia con más frecuencia a la DM1, pero hay que insistir en el control rutinario de marcadores de enfermedad celíaca para así hacer un diagnóstico precoz.
Diabetes mitocondrial: caso clínico
A. Casquero Cossío, A. Martín, S. Hernández, F. Camba, M. Clemente y M. Gussinyer
Servicio de Endocrinología Pediátrica. Hospital Materno-Infantil. Vall d'Hebron. Barcelona. España.
Antecedentes y objetivos: La diabetes mellitus se debe, en raras ocasiones, a defectos de los genes mitocondriales, asociando a veces sordera, o hepatopatía. Queremos destacar la importancia de pensar en esta entidad como un posible diagnóstico ante la presencia de hipertransaminemia más hiperglucemia.
Descripción clínica: Paciente de 17 años que consulta por hiperglucemia. Antecedentes personales: Recién nacido de 35 semanas, de 1.950 g al nacer. A los 5 años presenta elevación discreta de las transaminasas, virus hepatotropos (A, B, C, D) negativos, con ecografía abdominal normal. A los 12 años continúa presentando elevación de transaminasas (AST 110 U/l, ALT 139 U/l, GGT 70 U/l), sin otra alteración clínico-analítica. Es remitido al servicio de gastroenterología, donde resultan normales las siguientes exploraciones: analítica general (bilirrubina, coagulación, glucemia, creatincinasa 146 U/l,), serologías víricas (VHA, VHC, VHC, VHE, CMV, EBV), cupremia, ceruloplasmina, α 1-antitripsina, inmunoglobulinas, marcadores de celiaquía, marcadores de hepatitis autoinmune, test del sudor. Se realiza biopsia hepática por punción que se informa como normal (sin signos de histólisis). A los 15 años persiste hipertransaminemia y se realiza estudio metabólico. Ácidos orgánicos en orina: normal; azúcares reductores en orina: negativos; aminoácidos plasmáticos: normal; ácidos grasos libres: normal; estado redox plasmático: lactato 2,10 mmol/l; cupremia: 160 μg/dl; cupruria: 11 mg/l. Se detecta hipercolesterolemia de 249 mg/dl e hiperglucemia de 127 mg/dl. A los 16,5 años, con el fin de descartar alteraciones de la vía respiratoria, se realiza test de sobrecarga oral de glucosa: glucemia (120 min) 223 mg/dl, insulinemias inferiores a 30 μU/ml, lactato 3,5 mmol/l (60 min), lactato 3,20 mmol/l (120 min). Enfermedad actual: Paciente de 17 años remitido del servicio de Gastroenterología para valoración de hiperglucemia asintomática detectada en test de sobrecarga oral de glucosa. Exploraciones complementarias: Monitorización continua de glucosa (CGMS) en tres ocasiones: glucemias entre 150-200 mg/dl durante la mitad del registro, glucemia máxima posprandial de 270 mg/dl, HbA1c: 5,8 %. ENG y EMG: no alteración muscular ni de nervio periférico. Potenciales evocados auditivos: normales. Estudio genético (ADN mitocondrial): pendiente. Orientación diagnóstica: La presencia de hiperlactacidemia, insulinopenia e hipertransaminemia orienta al diagnóstico de diabetes mitocondrial.
Conclusiones: La diabetes mitocondrial es una entidad rara, pero se debe pensar en ella ante cuadros larvados de hiperglucemia, acompañados de otras manifestaciones, como elevación de transaminasas o alteraciones de la audición.
Diabetes mellitus neonatal probablemente permanente
A. Gutiérrez Macías, A. Escribano Muñoz, J.M. Martos Tello, M.J. Romero Egea y J.M. Donate Legaz
Servicio de Pediatría. Hospital Universitario Virgen de la Arrixaca. Murcia. España.
Introducción: La diabetes mellitus neonatal (DMN) se define como un cuadro de hiperglucemia durante el primer mes de vida, con duración mínima de 2 semanas y que requiere tratamiento insulínico para su control. Su incidencia es muy variable; se cifra en 1/400.000 recién nacidos vivos, y en nuestro país desconocida la incidencia posiblemente porque los casos nuevos no son publicados. La DMN presenta dos formas clínicas bien definidas por su evolución, que son prácticamente imposibles de delimitar clínica o biológicamente, en transitoria y permanente aunque si existen diferencias moleculares. Publicamos un caso de DMN posiblemente permanente dada la negatividad de su estudio molecular y su evolución de comienzo dramático.
Caso clínico: Varón hijo único de un matrimonio joven, no consanguíneo y sin antecedentes de diabetes ni de enfermedad autoinmune. Padre hepatitis A pasada. Gestación sin incidencias de 35 semanas. Parto con amniorrexis intraparto, de líquido claro, presentación cefálica, no instrumentada, Apgar 9/10 PN: 2.800 g. Acude al hospital a los 15 días, remitido por su pediatra, por estrés respiratorio y sospecha de deshidratación. Tos seca e intermitente con rinorrea de 3-4 días de evolución y rechazo del alimento las últimas 24 h. En el hospital los exámenes complementarios muestran glucemia, 1,161 mg/dl; pH 6,7, PCO2 25; bicarbonato 3,5, glucosuria y cetonuria. Se diagnostica de cetoacidosis diabética y se remite a hospital terciario con perfusión de bicarbonato. A su llegada impresiona de gravedad, presenta aspecto desnutrido y deshidratado con mucosa oral seca, signo del pliegue +, coloración pálida grisácea. Cráneo normoconfigurado con fontanela a punta de dedo deprimida, tórax simétrico con respiración acidótica, sin estertores y buena ventilación bilateral. Neurológicamente; reflejos arcaicos débiles, hipertonía de las cuatro extremidades, más marcada en miembros inferiores adoptando piernas cruzadas en tijera; pupilas mióticas. El resto de la exploración es normal. Bioquímicamente se constata la hiperglucemia, acidosis e hiperpotasemia (7,3), y son normales resto de iones, pruebas de coagulación, creatinina, PCR y radiografía de tórax.
Tratamiento: Tras la reexpansión de bicarbonato se inicia pauta de corrección de deshidratación clínica grave, estimada en un 10 %. A las 2 h del inicio se instaura perfusión de insulina a 0,05 U/kg/h precisando aumento máxima hasta 0,06 U/kg/h. Progresivamente se logra la corrección de la acidosis, así como el descenso gradual de los valores de glucemia y se mantiene perfusión de insulina durante 72 h. Posteriormente, se comenzó con insulina rápida i.v. en bolos tras cada toma y a las 72 h se comienza con insulina Lispro en pauta de multidosis con una dosis media de 0,6 U por toma (6 tomas diarias). Posteriormente, se instauró insulina NPH, con lo que se mantiene régimen (2 dosis de NPH e insulina Lispro a las 7 y 19 h y dosis, según glucemia, y Lispro antes de las tomas) con los que se consiguen glucemias correctas. Con 3 meses de vida se ha iniciado terapia con bomba de insulina subcutánea modelo Paradign 512 de Medtronic.
Resto exámenes complementarios: Insulina basal: 16 nU/ml, péptido C: 0,1 ng/ml, HLA: DR1,4//DQ5,8; ICA,() Anti-GAD, IAA, IA2 no enviados, anticuerpos antitiroideos negativos; T4, TSH normales, ecografías abdominal y cerebral normales; serie ósea normal; estudio cardiológico CIV muscular sin repercusión hemodinámica. El estudio genético y molecular realizado en el grupo GEDINE (Grupo de Investigación en Endocrinología y diabetes) del Hospital de Cruces, Vizcaya, es negativo.
Comentarios: Se realiza discusión sobre las distintas variaciones genéticas y moleculares entre DN permanente o transitoria. Se discute las dificultades del tratamiento en la DMN y de las armas terapéuticas de que disponemos: bomba de perfusión y tolbutamida.
Implicación de los canales de potasio en la diabetes neonatal
G. Pérez de Nanclares Leal 1, I. Estalella Bellart 1, C. Luzuriaga Tomás 2, J.R. Bilbao Catalá 1, I. Rica Etxebarria 1, L. Castaño González 1 y grupo GEDINE (Grupo Español para el estudio de Diabetes Neonatal)
Unidad de Investigación en Endocrinología y Diabetes. Endocrinología Infantil. 1Hospital de Cruces. Barakaldo. Bilbao. 2Hospital Marqués de Valdecilla. Santander. España.
Introducción: La diabetes neonatal se caracteriza clínicamente por la aparición de hiperglucemia durante los primeros 3 meses de vida. Existen dos formas de esta enfermedad: la diabetes neonatal transitoria, en la que la glucemia se normaliza y el tratamiento con insulina es necesario sólo durante unas semanas, y la diabetes neonatal permanente, en la que el tratamiento es de por vida. Los estudios genéticos realizados en los últimos años han asociado la enfermedad a alteraciones en la banda 6q24 (isodisomía paterna, duplicación de la banda 6q24 paterna o pérdida de metilación de la banda materna) y a mutaciones en los genes IPF1 y GCK. Aun así, estas alteraciones explicaban un porcentaje pequeño de los casos, hasta que se ha visto muy recientemente que existe otro gen asociado, el Kir6.2 que parece explicar muchas de las diabetes neonatales recogidas y analizadas por otros grupos.
Objetivo: El objetivo de este trabajo fue caracterizar las diabetes neonatales recogidas en nuestro grupo para lo cual se analizaron la región 6q24 y los genes IPF1, GCK y Kir6.2.
Pacientes y métodos: Se analizaron en total 18 familias independientes con al menos un miembro diagnosticado de diabetes neonatal. La isodisomía y duplicación de la banda paterna se estudió mediante microsatélites y la pérdida de metilación mediante enzimas de restricción y posterior amplificación por PCR, mientras que para la búsqueda de mutaciones en los genes IPF1, GCK y Kir6.2 se utilizó la secuenciación automática directa.
Resultados: De las 18 familias, una de ellas presentaba mutación en heterocigosis en el gen GCK (5,5 %), tres alteración de la banda 6q24 (16,6 %) y 8 mutación en el gen Kir6.2 (44,4 %). No se ha encontrado alteración en ninguna familia del gen IPF1 (0 %).
Conclusiones: 1. También en nuestro grupo las mutaciones en el gen Kir6.2 parecen explicar la mayoría de los casos de diabetes neonatal, aunque un 16,6 % son explicadas por alteraciones en la banda 6q24. 2. Mutaciones en heterocigosis del gen GCK, asociadas a la diabetes tipo MODY2, pueden dar lugar a una hiperglucemia mantenida durante el primer mes de vida y dar lugar por tanto a un diagnóstico de diabetes neonatal.
¿Es conflictivo el paso del adolescente diabético a la consulta de adultos?
F. Calvo Rigual, J. Galindo Grau, A. Merchante Alfaro, A. Gómez Sanz y D. Just Cardona
Servicio de Pediatría. Hospital Lluís Alcanyís. Xàtiva. Valencia. España.
Introducción: No existe consenso sobre la manera de realizar el paso del adolescente desde la consulta de endocrinología pediátrica a la consulta de adultos.
Objetivo: Demostrar que la transferencia no es problemática con un protocolo establecido, a una edad inferior de la habitualmente recomendada.
Material y métodos: Un total de 14 adolescentes diabéticos de un hospital comarcal que fueron transferidos desde la consulta de endocrinología pediátrica a la consulta de adultos mediante un protocolo preestablecido, a una edad entre 15 y 16 años, tras la creación de una Unidad de Diabetes que incluía ambas consultas. Se analiza la evolución de la enfermedad antes y después de la transferencia mediante la comparación de la hemoglobina glucosilada, índice de masa corporal, requerimientos de insulina, número de ingresos por complicaciones agudas, y cumplimiento de las visitas programadas. Se realiza un test de calidad de vida uno a dos años después de la transferencia.
Resultados: La media de duración de la diabetes en el momento de la transferencia fue de 6,85 años. La media de la hemoglobina glucosilada fue de 8,3 % antes de la transferencia, y de 8,66 % tras ella (no diferencia significativa). No hubo diferencias significativas en el índice de masa corporal (21,84 y 22,17 kg/m 2), ni en los requerimientos de insulina (0,83 U/kg/día y 0,80 U/kg/ día), antes y después de la transferencia. No hubo ingresos por complicaciones agudas en el período estudiado. La encuesta realizada tras la transferencia muestra unos resultados aceptables de calidad de vida.
Conclusión: La transferencia de adolescentes diabéticos en nuestro medio, mediante un protocolo establecido, no supone un empeoramiento del control y puede realizarse a una edad inferior de la recomendada en diversas publicaciones, pero más acorde con la normativa de nuestro sistema sanitario.
Peso neonatal en diabetes tipo MODY 2
I. Rica Etxebarria, I. Estalella, L. Castaño y Grupo español de Diabetes MODY
Servicio de Endocrinología Infantil. Unidad de Investigación. Hospital de Cruces. Barakaldo. Bilbao. España.
Introducción: La glucocinasa (GK) es una enzima presente en la célula β pancreática cuya función es decisiva para mantener la correcta homeostasis de la glucemia. Actúa como un sensor de la célula β a la glucosa regulando la síntesis y secreción de insulina en función de la misma. La diabetes MODY tipo 2 está originada por mutaciones inactivantes en heterocigosis del gen de la glucocinasa (cromosoma 7p). Mutaciones inactivantes en homocigosis del mismo gen son responsables de algunos casos de diabetes neonatal permanente. La insulina es una hormona clave en la regulación del crecimiento fetal. Por ello, los pacientes afectados de diabetes neonatal tienen un marcado retraso de crecimiento intrauterino, con mayor afectación del peso.
Objetivos: Estudiar la posible repercusión de la diabetes MODY 2 en el peso neonatal (PN). Comparar el PN de pacientes afectados de diabetes MODY 2 con el de sus hermanos sanos.
Pacientes y metodología: Hemos estudiado 73 familias diagnosticadas genéticamente de diabetes tipo MODY 2. La edad media del probando al diagnóstico fue de: 9,5 ± 5,4 años. El diagnóstico clínico de los pacientes fue diabetes 42 %, intolerancia hidrocarbonada 43 % e hiperglucemia 13 %. En el 75 % de ellos se había realizado sobrecarga oral de glucosa para el diagnóstico. El 81 % de los pacientes estaban tratados de forma exclusiva con dieta y el resto no recibían ninguna terapia. Al estudiar a las familias completas encontramos 30 hermanos afectados y 19 hermanos sanos.
Resultados: La media del PN de los pacientes afectados de diabetes MODY 2 está situada en el percentil 10 de nuestra población normal y es inferior al de sus hermanos sanos (2.992 ± 465 g frente a 3.627 ± 643). Esta diferencia se mantiene al comparar el PN de las 19 parejas de hermanos afectados y sanos (2.971 ± 564 g frente a 3.451 ± 635 g; test Wilcoxon; p = 0,003). El PN no varía en función de que la enfermedad se herede del padre o de la madre.
Conclusiones: Los pacientes con diabetes MODY tipo 2 tienen un PN disminuido y menor que sus hermanos sanos. Este hecho es atribuible a una disminución parcial de la secreción fetal de insulina intraútero como consecuencia de la mutación en el gen de la glucocinasa.
Diabetes mellitus en paciente afectada de fenilcetonuria
J. Bosch Muñoz, M. García García, C. Martín Ansón, M. Plana, Z. Ibars y A.R. Gomà Brufau
Servicio de Pediatría. Hospital Arnau De Vilanova. Lleida. España.
Introducción: La fenilcetonuria es un trastorno metabólico que asocia retraso mental y rasgos fenotípicos especiales. Si se diagnostica precozmente y se comienza con una dieta baja en fenilalanina no presentan las complicaciones típicas de la enfermedad. La diabetes mellitus insulindependiente es otra enfermedad metabólica en la que además del aporte exógeno de insulina una de las bases del tratamiento es una dieta adecuada.
Caso clínico: Niña de 11 años, fruto de una segunda gestación controlada y tolerada de 34 semanas gestacionales por rotura prematura de membranas. Nace con un peso de 2.180 g y una longitud de 45,5 cm. Entre los antecedentes familiares destaca que su hermana de 2 años, presenta fenilcetonuria clásica. Por este motivo se recoge muestra para valoración de fenilalaninemia. En la primera determinación se obtiene una cifra de 1.100 mmol/l. En ese momento se cambió su dieta a una fórmula libre de fenilalanina y se remitió a consultas del centro de referencia. El seguimiento de esta paciente ha permitido poner de manifiesto un desarrollo neurológico y psicomotor normal. El rendimiento escolar debe considerarse óptimo. El estudio de las funciones cognitivas es normal, excepto en las de tipo ejecutivo, en las que presenta ciertas dificultades en la estrategia de resolución de problemas. Se ha practicado densitometría y se aprecia osteopenia, por la que recibe tratamiento con suplementos de calcio. Sigue controles en la unidad de fenilcetonuria y mantiene un buen control dietético y estabilidad en los niveles de fenilalanina debido a que ha mantenido en estos años una alimentación ajustada a las recomendaciones confeccionadas para ella los dietistas. A los 9 años presentó cuadro de poliuria, polidipsia y pérdida de peso de un mes de evolución. Tras la aparición de dolor abdominal consultan con su pediatra, que diagnostica el proceso de inicio diabético al obtener una glucemia de 286 mg/dl. Es ingresada en nuestro centro para iniciar tratamiento. A su ingreso presenta buen estado general y se estima una deshidratación del 3 %. Peso de 27,5 kg, talla de 135 cm (P25). El resto de la exploración física fue normal. Se objetiva una glucemia de 553 mg/dl, con un pH 7,27, bicarbonato 16,04 y exceso de base de 9,7, creatinina 1,21 mg/dl, urea 34 mg/dl, sodio 134,5 mEql/l, glucosuria y cetonuria, HbA1c 10,7 %. Se instaura tratamiento habitual se obtiene una buena respuesta metabólica y adecuada adaptación tanto a la dieta como a la administración de insulina. Durante su estancia hospitalaria requiere dosis de 0,6 U/kg que en el momento del alta es de 0,25 U/kg. Continúa seguimiento en consultas externas de endocrinología infantil con un excelente control metabólico, teniendo una HbA1c que oscila entre 6,6 y 7,2 % (VN 4,6-6). En la actualidad precisa una dosis de insulina de 0,7 U/kg. Ha iniciado el desarrollo puberal estando en estadio II de Tanner.
Comentarios: El tratamiento de la fenilcetonuria es nutricional. Debe tenerse en cuenta: el diagnóstico precoz en los primeros días de vida, tratamiento durante toda la vida, control intensivo durante la gestación y si es posible en un período previo al inicio del embarazo y la restricción de fenilalanina para mantener concentración inferior a 360 μmol/l en sangre. La dieta seguida por la paciente a partir del momento en que se conoce la enfermedad diabética no ha presentado prácticamente ninguna variación respecto a la que consumía antes del inicio diabético. Su estado nutricional se ha mantenido en niveles adecuados y no se ha objetivado ningún cambio significativo en el desarrollo de sus hábitos. El hecho de estar acostumbrada a un régimen alimenticio no le ha generado dificultades para adaptarse a las pequeñas variaciones en su alimentación, dieta por raciones de hidratos de carbono, ya que a lo largo de toda su vida ha seguido una dieta muy vigilada.
Revisión de los casos de inicio diabético en Lleida en el período 1995-2004 en niños hasta 14 años
J. Bosch Muñoz, C. Martín Ansón, M. García García, M. Plana, Z. Ibars y A.R. Gomà
Servicio de Pediatría. Hospital Universitari Arnau de Vilanova. Lleida. España.
Introducción: La incidencia de la diabetes mellitus tipo 1 en la edad pediátrica oscila notablemente entre los distintos estudios publicados. Las variaciones se observan entre las distintas áreas del globo terrestre. Corea, China, Japón con cifras alrededor del 1 por 100.000 presentan los valores más bajos, mientras que Finlandia tiene una incidencia de 35 por 100.000. En Europa, es conocida la existencia de un gradiente Norte-Sur, con mayor índice en las zonas septentrionales, aunque con salvedades, como es el caso de la isla de Cerdeña, cuyos valores son muy superiores a los de las áreas próximas. Al margen de la amplia variación geográfica mundial, se ha observado en varios países en aumento continuado de la incidencia de la enfermedad a lo largo de los últimos 10 años.
En España los distintos análisis también muestran variaciones regionales, aunque se ha visto que no es tan claro el gradiente Norte-Sur. En Cataluña, conocemos las cifras actuales, gracias a la existencia del Registro de Diabetes tipo 1 en activo desde el año 1987. En este contexto, hemos realizado un análisis retrospectivo de la incidencia en nuestra provincia. Lleida tiene una superficie de 12.172 km 2, con una población referida al censo de población del año 2001, (intermedio en nuestro estudio) de 48.305 habitantes. Corresponden al sexo masculino una población de 24.678 y al sexo femenino de 23.627 con una distribución por edades de 0 a 4 de 16.054 habitantes, de 5-9 de 15.455 y de 10 a 14 de 16.796. Nuestro hospital, único referente en endocrinología infantil en la provincia atiende a dicha población y además a la de la zona geográfica denominada Franja de Ponent, que corresponde a Aragón (provincias de Huesca y Zaragoza). La atención sanitaria a esta población está recogida en los acuerdos sanitarios entre ambas comunidades. Esta población está compuesta según el mismo censo por un total de 7.798 habitantes. Corresponden al sexo masculino 4.020 habitantes y al femenino 3.778, con una distribución por edades de 0 a 4 años 2.535 habitantes, de 5 a 9 años de 2.581 y de 10 a 14 años de 2.682.
Resultados: Se han recogido 81 casos de inicio diabético. 46 niños y 35 niñas con una relación entre sexos de 1,3 a favor del masculino. La incidencia global en este período es de 14,43. Por sexos obtenemos un valor de 16,02 en el masculino y de 12,77 en el femenino. Por grupos de edad, de 0 a 4 años la incidencia es de 8,6, en el de 5 a 9 años la cifra corresponde a 12,75 y en el de 10 a 14 años obtenemos un valor de 21,56. La cetoacidosis diabética en el momento del inicio corresponde a un 45,6 % del total con una distribución por sexos de 75,6 % en niños y de 24,32 en niñas. Los valores de HbA1c en el ingreso oscilan entre 10,5 y 14,7 % con una media de 11,5 %. Como enfermedades autoinmunes asociadas obtenemos cuatro casos de enfermedad celíaca y dos de tiroiditis. Los porcentajes de inicio en función de la estación del año son primavera 29,6; verano 20,9; otoño 23,45, e invierno 25,92.
Comentarios: Los datos correspondientes a Cataluña, referente al período 1990-1994 mostraron una incidencia de 12,3 por 100.000 en niños menores de 15 años. En un estudio retrospectivo realizado en nuestro hospital en los años 1982-1994, la cifra obtenida fue de 10,33 por 100.000, que es semejante a la cifra total de Cataluña. Las cifras obtenidas en este estudio muestran un ligero incremento respecto al período 1982-1994, lo cual es congruente con los datos publicados por otros autores. Apreciamos un incremento en el grupo de 10 a 14 años y creemos que seguimos teniendo una cifra elevada de inicios con cetoacidosis.
Cetoacidosis diabética y rabdomiólisis
J. Barreiro Conde, T. Arévalo, M. López, F. Martinón, L. Iglesias, F. Vasconcelos Espada, P. Cabanas, L. Castro Feijoó y M. Pombo
Hospital Clínico y Universidad de Santiago de Compostela. España.
Introducción: La cetoacidosis diabética (CD) es una complicación aguda de la diabetes mellitus tipo 1 y está asociada a numerosos desequilibrios biológicos. Además de la tríada clásica (hiperglucemia, cetonuria y acidosis metabólica) en algunos pacientes se puede encontrar otras anormalidades enzimáticas. La CD puede presentar varias complicaciones como: pancreatitis, edema cerebral (la más frecuente) y rabdomiólisis, pues hay pocos casos descritos de esta última en la población infantil.
Caso clínico: Niña de 11 meses que consulta por dificultad respiratoria y somnolencia. Antecedentes personales: Tercer embarazo de alto riesgo por desprendimiento de placenta, dos abortos espontáneos previos; parto a las 38 semanas, peso de recién nacido de 2.560 g. Período neonatal sin incidencias. Lactancia materna exclusiva hasta el quinto mes de vida. Posteriormente añaden fruta y verduras, pero no carne, pescado, ni cereales. Sin vacunación. Antecedentes familiares: Padres homeópatas y vegetarianos. Sin historia familiar de diabetes ni enfermedades autoinmunes. Anamnesis: Una semana de evolución de nicturia, polidipsia, polifagia, pérdida de peso y dificultad respiratoria 48 h previas a su ingreso. Exploración física: Peso 6,2 kg; longitud 73,5 cm (P50). PA: 90/40 mmHg. Afectación del estado general. Glasgow 12. Signos de deshidratación grave. Resto sin alteraciones destacables. Pruebas complementarias: glucemia 1.168 mg/dl; pH 6,96; pCO2 11,5 mmHg; EB 27,3 mmol/l; bicarbonato 5,3 mmol/l; urea 88 mg/dl; creatinina 1,3 mg/dl; CPK 122 U/l (VN: 1-110) con iones normales. Hb 10,5 g/dl; Hto 34,7 %; plaquetas 221.000/μl; cetonemia > 6 mmol/l. Tratamiento y evolución: Ante el diagnóstico de diabetes mellitus tipo 1 con cetoacidosis severa se aplica el protocolo de consenso de la Sociedad Europea de Endocrinología Pediátrica con buena evolución. A las 24 h: pH 7,28; EB 15,3 mmol/l; glucemia 298 mg/dl, y destaca irritabilidad (TC craneal no edema cerebral). En control analítico: CPK 2,530 U/l; mioglobina 330 ng/ml; troponina < 0,04 U/l. Con el diagnóstico de rabdomiólisis se procede a hiperhidratación (3 l/m 2/día) y alcalinización urinaria con mejoría paulatina de la clínica y de los valores de CPK. Progresivamente, se aprecia un descenso de plaquetas (25.000/μl) y hemoglobina (6,9 g/dl), que precisa transfusión de concentrado de hematíes. Serologías virales negativas, hemocultivo negativo y se aísla en urocultivo Escherichia coli (> 100.000 colonias/ml). Se inicia tratamiento con antibioterapia i.v.
Comentarios: La patogenia de la rabdomiólisis en la cetoacidosis diabética es incierta. Se ha asociado a valores altos de glucemia, alteraciones electrolíticas y osmolares. En toda CD es necesario evaluar otras alteraciones metabólicas y tratarlas precozmente para disminuir la morbimortalidad.
Relación entre el sistema HLA y la diabetes mellitus tipo 1: sus variaciones en los diferentes países y poblaciones
S. Conde Barreiro 1, G.M. Lou Francés 1, E. Javierre Miranda 1, I. Echevarría Matía 1, M. Rodríguez Rigual 1 y J.M. Garagorri Otero 2
1Servicio de Endocrinología Pediátrica. Hospital Infantil Miguel Servet. 2Hospital Clínico Universitario Lozano Blesa. Zaragoza. España.
Introducción: Los conocimientos acerca de la predisposición genética en la diabetes mellitus tipo 1 (DM1) se han multiplicado en los últimos años. Numerosos estudios han demostrado la importancia del sistema HLA (locus IDDM1), y se han estudiado múltiples alelos y combinaciones de los mismos que confieren protección o susceptibilidad a la DM1.
Objetivos: Revisar los trabajos publicados en los últimos años que estudian la relación entre el HLA y la DM1 en pacientes pediátricos y contrastar los resultados obtenidos en los distintos países y poblaciones.
Material y métodos: Se ha utilizado el sistema PUBMED, para buscar los artículos que reunieran los términos diabetes mellitus, type 1 y HLA, acotando la búsqueda a los trabajos referidos a menores de 18 años, y restringiendo la fecha de publicación a los últimos 12 años (desde junio de 1993). Así encontramos más de 500 artículos, que luego se han cribado según el título y el resumen de los mismos, y se han escogido sólo los que hacían referencia al propósito de este trabajo. Quedaron finalmente 206 estudios de todo el mundo, en los que la lectura del abstract y/o las conclusiones ha bastado en casi todos los casos para conocer sus resultados. Se han agrupado los diversos estudios por regiones geográficas (continentes y países) para mostrar los resultados en cada una de ellas.
Resultados: Los países con más trabajos publicados fueron China, Finlandia, Estados Unidos, Italia y Suecia. La mayor parte de los trabajos estudian la distribución de los distintos alelos y haplotipos del sistema HLA en diabéticos en regiones geográficas o grupos étnicos concretos. También encontramos algunos estudios multicéntricos que tratan conjuntamente la distribución del HLA y la diabetes en varios países. Prácticamente en todas las poblaciones estudiadas se encuentra la presencia de determinados haplotipos de riesgo (DRB1*03-DQA1*0501-DQB1*0201 y DRB1*04-DQA1*0301-DQB1*0302 principalmente), así como de protección (DRB1*1501-DQA1*0102-DQB1*0602), si bien algunos estudios muestran la presencia de otros haplotipos más representados en determinadas poblaciones. Muchos estudios concluyen que la confluencia de varios alelos de riesgo en un mismo paciente dan lugar a formas de diabetes más precoces y agresivas, de mayor capacidad de producción de determinados autoanticuerpos propios de la DM1, e incluso de mayor riesgo de aparición de otras enfermedades autoinmunes. Algunos estudios revelan la existencia alelos predisponentes y protectores propios de poblaciones y etnias concretas.
Conclusiones: La investigación de la relación HLA/DM1 está especialmente desarrollada en países con alta incidencia de DM1, así como en países en que su incidencia varía mucho entre unas regiones y otras. Los estudios muestran la presencia de diversos alelos y haplotipos HLA que son comunes a múltiples poblaciones en todo el mundo, y que tienen un claro efecto predisponente o protector frente a la DM1. Se pueden identificar variaciones que afectan directamente a las diferencias de incidencia y forma de presentación de la DM1 entre distintas poblaciones, si bien el sistema HLA no explica totalmente estas diferencias, así que es necesario seguir investigando otros factores genéticos y ambientales que influyen también en la patogenia de la DM1.
Satisfacción con el uso de un sistema de autodiagnóstico de glucemia y cuerpos cetónicos (sistema Optium) en pediatría
F. Hermoso López 1, P. Bahillo Curieses 1 y M. Parramón Ponz 2
Departamento de Pediatría. Unidad de Endocrinología. 1Hospital Clínico de Valladolid. 2Abbott Científica. España.
Introducción: La valoración de los padres y del personal sanitario de los dispositivos de autodiagnóstico que están disponibles en el mercado para el control de la diabetes no se suele realizar, por este motivo no se hacen públicas estas opiniones. Cuantificar la percepción de las distintas características y/o prestaciones puede ayudar hacer un juicio a la hora de conocer las opiniones de los clínicos y de sus pacientes. Además, puede contribuir a la educación en el autocontrol de la diabetes.
Objetivos: Valorar el grado de satisfacción de los usuarios de un dispositivo de autodiagnóstico de glucemia y cuerpos cetónicos para diabetes (sistema Optium®). Comparar el grado de control que supone la determinación de dos parámetros a lo largo del tiempo de seguimiento en relación con un grupo control en el que sólo se valora la glucemia.
Pacientes y métodos: Se incluyeron en el grupo de estudio 20 niños de 9,13 ± 4,23 años (2-18) con diabetes mellitus tipo 1 de 3. 87 ± 3,18 años de evolución [0,35-12]. La variable principal estudiada fue la puntuación del cuestionario de satisfacción (escala del 1 al 5) específico del uso de un dispositivo de autodiagnóstico (MediSense Optium®), y sus tiras de glucemia (MediSense Optium® Plus, 10 s, 1,5 μl) y cuerpos cetónicos (MediSense Optium® beta-ketone, 30 s, 5 μl). Las variables secundarias fueron el grado de control de la diabetes y de las glucemias por los padres y por el centro (puntuaciones del 1 al 10), el número de visitas inesperadas, el número de cetosis o cetoacidosis registradas durante 2 meses de seguimiento. Los niños se medían los cuerpos cetónicos en sangre capilar si en la mañana tenían glucemias de más de 250 mg/dl. El resto del día, se efectuaba un control glucémico normal, con la excepción de circunstancias especiales como trasiego dietético, enfermedad concomitante, etc. Las variables secundarias se compararon respecto a un grupo control, formado por 20 niños que iban a la misma consulta y usaban cualquier otro medidor, durante el mismo tiempo de seguimiento que el grupo de estudio. El criterio de cetosis fue β-hidroxibutirato capilar igual o superior a 0,6 mmol/l. El criterio de cetoacidosis era la existencia de sintomatología clínica evidente como respiración acidótica o deshidratación. Los grupos no se hicieron por selección, sino por orden de llegada. Se hizo la estadística descriptiva de las variables demográficas y se calcularon las medias de las puntuaciones obtenidas, con SPSS 11,5.
Resultados: El grado de satisfacción en una escala del 1 al 5 fue: para el medidor, 4,22; volumen y aplicación de la tira de cuerpos cetónicos (depósito de gota), 3,85 y 3,52, respectivamente; utilidad de la tira de cuerpos cetónicos, 4,95; valoración de medidor y tiras en su conjunto, 4,15. La media global de todas las puntuaciones fue 4,18. Con puntuaciones del 1 al 10, el grado de seguridad en el control de la diabetes tanto por los padres como por el personal sanitario fue de 8,7 en el grupo de Optium®, frente a 7,9 en el grupo control. El grado de seguridad en el control de la glucemia por los padres fue de 8,6 y por el centro 8,5 en el grupo de Optium® frente a 7,9 en el grupo control. En ninguno de los grupos se registraron visitas inesperadas, hipoglucemias graves, acontecimientos adversos o cetoacidosis durante el estudio. Se registraron 28 cetosis en el grupo control y 16 cetosis en el grupo de Optium®.
Conclusiones: El uso de un cuestionario específico ha permitido valorar el grado de satisfacción y seguridad que ofrece un medidor de glucemia y cuerpos cetónicos en sangre capilar, ésta es muy alta (4,22 sobre 5), y se recomendaría el uso de este sistema a otras personas (4,6 sobre 5). En especial, la medida de cuerpos cetónicos en sangre es muy apreciada por los padres (4,95 sobre 5). La elevada media global de todas las puntuaciones de satisfacción, así como el grado de seguridad con el control de la diabetes permitieron deducir que el sistema Optium®, con las tiras Optium plus® beta-ketone, es muy apreciado tanto por los padres como por el personal sanitario.
Síndrome hiperosmolar hiperglucémico no cetósico
L. Sentchordi Montané, A. Aragonés Gallego, B. Brandstrup Azuero, A. Crespo Alonso, R. Tapia Moreno y M.D. Sánchez-Redondo
Hospital Virgen de la Salud. Toledo. España.
Introducción: El síndrome hiperglucémico hiperosmolar se caracteriza por una fuerte hiperglucemia (glucosa > 600 mg/dl), una escasa acidemia (pH > 7,3 HCO3 > 15 mEq/l), una profunda deshidratación (osmolaridad sérica > 320 mOsm/kg) en ausencia de acidosis y grados variables de alteración del sensorio, desde la obnubilación al coma. Es una patología con elevada morbimortalidad que aparece en pacientes ancianos con DMNID. Por el contrario, es rara en niños, y se han descrito pocos casos en la literatura médica, aunque recientemente están apareciendo en relación con DM tipo 2 inadvertidas previamente. La forma de presentación es insidiosa, debido a la ausencia de acidosis. Presentamos un caso en un varón de 8 años con DM tipo 1 de un año de evolución que presenta un episodio de estas características.
Caso clínico: Paciente varón de 7 años y 9 meses que acude a urgencias por glucemias capilares superiores a 500 mg/dl en las últimas 48 h, acompañadas de poliuria y polidipsia. Exploración física: P: 23 kg (P25), T: 128 cm (P75), T.ª: 36,5 °C, FC: 126 lat./min, FR: 16 rpm, PA: 118/65 mmHg. Aceptable estado general, decaído. Buen color de piel y mucosas. Mucosa oral seca, lengua pastosa, no fetor cetonémico. ACP: normal. Abdomen: normal. ORL: normal. Ex. neurológico: Glasgow 13-14, desorientado y con tendencia al sueño. Exámenes complementarios: Gasometría venosa: pH 7,30, pCO2 40,1 mmHg, Bic 19,4 mEq/l, EB 5,7 mm/l. Hemograma: normal. Bioquímica: glucosa 914 mg/dl, urea 46 mg/dl, creatinina 0,4 mg/dl, Na 141 mEq/l, K 5 mEq/l, Cl 90 mEq/l, Mg 2,11 mg/dl, osmolaridad: 332 mOsm/kg. S. de orina: densidad 1005, glucosuria ++++, cetonuria+. Evolución: Ingresa en UCIP donde recibe fluidoterapia i.v. así como perfusión de insulina. Presenta disminución progresiva de glucemia y osmolaridad. Clínicamente mejoría rápida de la alteración de la conciencia. No se observaron complicaciones a corto ni largo plazo.
Comentarios: 1. El estado hiperosmolar no cetósico es una condición rara en pediatría, asociada con problemas psiquiátricos y familiares. 2. El tratamiento es similar al de la cetoacidosis diabética, y está más recomendada la utilización de fluidos hipotónicos tras la fase de estabilización y la menor necesidad de insulina. 3. La hiperosmolaridad, unida a la deshidratación favorece la aparición de complicaciones neurológicas.
Síndrome metabólico: observaciones en 22 pacientes
P. Prieto Matos, C. Criado Muriel, M. Mateos Polo, M. Martín y J. Prieto Veiga
Unidad de Endocrinología Pediátrica. Hospital Universitario de Salamanca. España.
Introducción: Incluimos bajo esta denominación a aquellos niños/as que presentan al menos dos de los siguientes síntomas: obesidad, HTA, resistencia a la insulina, dislipemia y que pueden asociar hiperandrogenismo, acantosis nigricans, etc.
Pacientes y métodos: Hemos seleccionado 23 pacientes que reúnen requisitos de síndrome metabólico de 42 diagnosticados de obesidad. Se valora IMC, cociente cintura-cadera (valores superiores a 0,9 en la niña y de uno en el niño fueron considerados elevados), dislipemia, HTA, cociente glucemia-insulina, clínica de precocidad sexual, acantosis nigricans.
Resultados: Obesidad: 23 pacientes tuvieron IMC superior a percentil 95. Cociente cintura/cadera elevados: 11 de los 22 pacientes. Dislipemia (aumento de colesterol total, c-LDL o disminución de c-HDL o aumento de triglicéridos o alteración de los índices c-HDL o LDL/HDL: 14 pacientes. Hipertensión arterial: 6. Resistencia a la insulina: 15. Acantosis nigricans: 5. Síntomas de hiperandrogenismo o precocidad sexual: 4. En los casos en que tras el tratamiento inicial de pérdida de peso se mantienen niveles elevados de insulina se asoció metformina.
Conclusión: La evaluación de la clínica asociada a la obesidad es de gran interés debido a que la suma de otros factores aumenta el riesgo de complicaciones. El tratamiento del SM debe abarcar el amplio espectro de patología asociada.
Comparación entre el tratamiento con insulina glargina e infusión continua de insulina en pacientes pediátricos con diabetes mellitus tipo 1
E. Lara Orejas, E. Colino, M. López-Capapé, Y. Alins, M. Martín, M. Alonso y R. Barrio
Pediatría. Unidad de diabetes pediátrica. Hospital Universitario Ramón y Cajal. Madrid. España.
Introducción: El estudio del DCCT demostró que el buen control metabólico previene o retrasa la aparición de las complicaciones microvasculares de la diabetes. Para conseguirlo, hoy en día, se dispone fundamentalmente de dos tipos de terapias: la terapia de múltiples dosis de insulina (MDI) y los dispositivos de infusión continua de insulina (BICI). Desde diciembre de 2003, en España, disponemos de la insulina glargina, primer análogo de acción prolongada. Esta insulina tiene una duración de 24 h, menor variabilidad de absorción y un perfil más estable que la NPH.
Objetivo: Comparar la eficacia de la terapia con MDI utilizando insulina glargina y BICI.
Material y métodos: Dividimos los pacientes en dos grupos: el primero (grupo A) incluye 19 pacientes tratados con BICI con análogo de acción rápida y el segundo (grupo B) con 19 pacientes tratados con MDI con insulina glargina y análogos de acción rápida. Ambos grupos tenían edad, índice de masa corporal (IMC), tiempo de evolución de la diabetes, necesidades de insulina y grado de control metabólico semejantes. En todos ellos valoramos al inicio del cambio y a los 6 meses las necesidades de insulina (U/kg/día), IMC, episodios de hipoglucemia grave, episodios de cetoacidosis y el grado de control metabólico mediante la determinación bimensual de HbA1c (HPLC Menarini, VN: 5,1 ± 0,31 %).
Resultados: La HbA1c mejoró en ambos grupos, disminuyendo de 8,29 ± 1,55 a 7,71 ± 0,92 (p < 0,05) a los 6 meses en el grupo A y de 7,89 ± 1,16 a 7,19 ± 0,72 (p < 0,005) en el grupo B. La dosis de insulina también disminuyó en ambos de forma significativa, en el tratado con BICI de 0,94 ± 0,27 a 0,8 ± 0,18 (p < 0,05) y en el grupo B de 0,92 ± 0,38 a 0,78 ± 0,31 (p < 0,005). No hubo diferencias significativas entre ambos grupos ni en la HbA1c ni en la dosis de insulina. Las hipoglucemias graves disminuyeron en ambos grupos pero no de forma significativa. El IMC no se modificó. Sólo un paciente tratado con BICI tuvo un episodio de cetoacidosis.
Conclusiones: 1. Se puede conseguir un control metabólico similar utilizando terapia con MDI con insulina glargina y análogo de acción rápida que con BICI en los pacientes pediátricos con diabetes tipo 1. 2. Ambas formas de terapia permiten mejorar el control metabólico sin incrementar el riesgo de complicaciones agudas. Es necesario un seguimiento a más largo plazo para verificar estos resultados. Debido al mayor coste económico del tratamiento con BICI recomendamos utilizar la terapia con MDI con glargina antes de considerar la necesidad del tratamiento con BICI.
Un año de tratamiento con glargina
A. González Vergaz, B. García Cuartero, M. Fernández y M.J. Obra
Pediatría. Hospital Severo Ochoa. Leganés. Madrid. España.
Introducción: Los análogos de insulina de acción prolongada son una opción terapéutica que pueden mejorar el control metabólico y la calidad de vida de los pacientes diabéticos.
Objetivo: Valorar la evolución clínica, analítica y de calidad de vida en un grupo de niños y adolescentes diabéticos al año de iniciar tratamiento con insulina glargina.
Pacientes y métodos: Un total de 30 pacientes con edades comprendidas entre 6,1 y 17 años (media 13,6) 18 niños y 12 mujeres, 17 puberales y 13 prepuberales, están incluidos en este estudio. El índice de masa corporal (IMC) excepto en 2 mujeres adolescentes se encontraba en la media ± una desviación estándar para su edad y sexo. Tres de los pacientes presentaban patología asociada (enfermedad celíaca). El tiempo medio de evolución de la diabetes era de 4,6 años (1,4-9,8 años) y todos ellos recibían tratamiento previo con terapia intensiva (análogo de acción rápida preprandial 3 o 4 veces al día y NPH 2-3 veces al día). La dosis total de insulina administrada era 0,88 ± 0,37 U/kg/día y correspondía el 32,9 % ± 11,8 al análogo de acción rápida. La media de las HbA1c del año previo era 7,34 % ± 0,95 (5,5-9,02 %). Los motivos del cambio a insulina glargina fueron los siguientes: mal control metabólico, gran irregularidad en las glucemias preprandiales, fenómeno del alba o necesidad de flexibilidad de horarios. Se inicia administración de insulina glargina por la noche en 23 pacientes y en 7 al desayuno. Se reduce la dosis el 15-20 % respecto a la insulina intermedia que se administraba, manteniendo el análogo de acción rápida. Tras 8 meses de tratamiento se solicita a los pacientes su opinión sobre su control metabólico y sobre la repercusión en su calidad de vida.
Resultados: Dos pacientes abandonaron tratamiento por hiperglucemia en el primer mes de tratamiento; 3 pacientes tras 6 meses, vuelven a NPH por incremento de HbA1c. Ningún paciente sufrió modificaciones significativas en el IMC. Dos pacientes requirieron ingreso en una ocasión por cetoacidosis tras 11 meses de tratamiento y ninguno presentó hipoglucemia severa. La administración de glargina al año de tratamiento se mantenía nocturna en 12 pacientes, tres en la comida y nueve en el desayuno. La dosis total de insulina al año de tratamiento fue muy similar a la previa: 0,84 ± 0,32 U/kg/día pero se incrementa el porcentaje de análogo de acción rápida 40,2 % ± 8,6. La media de las HbA1c en los pacientes que completaron el año de tratamiento fue 7,63 ± 1,1. Los datos de la encuesta reflejan que la mitad de los pacientes consideran que su control es más estable y presentan menos hipoglucemias; el 76 % que es más cómoda y el 68 % que mejora su calidad de vida y el 38 % opina que mejora su control metabólico.
Conclusiones: 1. No encontramos cambios en el IMC ni en la dosis total de insulina por administrar, aunque incrementan el número de veces que emplean el análogo de acción rápida y el porcentaje de este empleado. 2. No encontramos mejoría en la HbA1c. 3. Los pacientes puberales que precisan flexibilidad horaria y los prepuberales que comen en el colegio son los más satisfechos.
Valores de referencia de insulinemia, péptido C y HOMA en niños sanos mediante quimioluminiscencia
B. García Cuartero, C. García Lacalle, C. Jiménez Lobo, A. González Vergaz, E. Díaz y N.J. Alcázar
Servicio de Pediatría. Hospital Severo Ochoa. Leganés. Madrid. España.
Introducción: En los últimos años, dentro de la población infantil, se ha producido un incremento importante de algunas enfermedades metabólicas de gran repercusión social y económica como la diabetes mellitus, tanto tipo 1 como 2, y la obesidad debido a cambios en los hábitos de vida. Algunas de ellas son de riesgo para cuadros de insulinorresistencia y síndrome metabólico. La insulinemia, el péptido C en ayunas y el índice HOMA son parámetros básicos cuya determinación orienta el diagnóstico de estos pacientes, además de informar sobre la reserva pancreática. Por ello el objetivo del presente trabajo ha sido la estandarización de los valores de insulina, péptido C e índice HOMA relacionados con el IMC en nuestra población infantil sita en la comunidad autónoma de Madrid.
Metodología: Un total de 350 niños y adolescentes de ambos sexos y estadios puberales, sanos de la comunidad autónoma de Madrid participaron en el estudio tras consentimiento informado de los padres y del comité ético. Se determinaron los valores en ayunas de insulinemia y péptido C mediante quimioluminiscencia (ELECSYS; Roche), y se calculó el índice HOMA, así como el IMC: peso/talla 2. Para análisis estadístico se empleó el SPSS versión 11.0. Las variables se expresaron como media +DE. El análisis de la varianza mediante Kolmorogov, Shapiro.Wilk, t de Student, U de Mann. Whitney, Bonferroni, Kruskall-Wallis y test de Levene. Nivel de significación p < 0,05.
Resultados: Se especifican en las figuras 1, 2 y 3.
Figura 1.
Figura 2.
Figura 3.
Conclusiones: Los valores de insulinemia, péptido C e índice HOMA aumentan con la edad y sobre todo con el estadio puberal para ambos sexos. No se encontraron diferencias significativas para cada uno de los parámetros entre sexos. La obtención de estos valores de referencia en nuestra población, dado el aumento de la prevalencia de ciertas patologías de riesgo cardiovascular, nos será de gran utilidad para un mejor diagnóstico clínico de nuestros pacientes.
Análisis de la fiabilidad de dos dosímetros portátiles de glucemia
S.I. De la Torre Santos, J.M. Andrés de Llano, R. Cancho Candela y J.E. Fernández Alonso
Servicio de Pediatría. Complejo Hospitalario de Palencia. España.
Introducción: La determinación de la glucemia es un parámetro de uso habitual en la práctica clínica. Se ha cuestionado la fiabilidad de los resultados obtenidos por dosímetros portátiles en comparación con los valores determinados en el laboratorio, sobre todo en situaciones de hipoglucemia.
Objetivo: Comparación de dos dosímetros portátiles en la medición de concentración de glucemia en recién nacidos.
Sujetos, material y métodos: Se trata de un estudio descriptivo transversal. La fiabilidad se evaluó mediante el coeficiente de correlación intraclase (CCI) y el análisis de Bland-Altman. Se analizaron 82 determinaciones en 65 niños. Se realizaron determinaciones en sangre capilar de talón y en vena periférica. Se usaron dos reflectómetros modelo Glucocard Memory® y un analizador central modelo Dimension-RxL.
Resultados: La distribución por sexo fue del 58,8 % varones y 41,3 % mujeres. La mediana de la edad es de un día, con una amplitud intercuartil de 5 días. La media del peso es 3.249,75 g, con una desviación estándar de 711,15 g. En las comparaciones realizadas mediante CCI entre las medidas obtenidas por el laboratorio y los dosímetros se encontraron valores entre 0,79-0,83 y en la comparación entre los dosímetros los valores fueron entre 0,78-0,99. En el análisis de Bland-Altman los intervalos de concordancia fueron más amplios cuando se comparó el analizador central frente a los dosímetros (40-42 mg/dl). En la comparación entre los dosímetros entre sí el intervalo de concordancia fue de 9-43 mg/dl.
Conclusiones: Las medidas de laboratorio infraestiman los valores de glucemia, probablemente por el retraso en el análisis de la muestra. Ambos reflectómetros son intercambiables entre sí desde el punto de vista clínico. Valores extremos de glucemia (hipoglucemia) no influyen en la validez.
Intolerancia a la glucosa en niños y adolescentes obesos. Relación con el grado de adiposidad
D. Yeste Fernández, R. Tomasini, G. Dodino, M. Clemente, A. Albisu, N. Potau, M. Gussinyé y A. Carrascosa
Servicio de Endocrinología Pediátrica. Hospital Materno-infantil Vall d'Hebron. Barcelona. España.
Introducción: El incremento de la prevalencia de la intolerancia a la glucosa y de la diabetes tipo 2 en la infancia y la adolescencia en las últimas décadas parece estar asociada al aumento de la incidencia de obesidad en las sociedades desarrolladas.
Objetivos: Conocer la frecuencia de la intolerancia a la glucosa y de la diabetes tipo 2 en una población de niños y adolescentes obesos evaluados en nuestro hospital y su relación con el grado de adiposidad.
Pacientes y métodos: Estudio prospectivo de 147 pacientes obesos (60 varones con IMC de 29,5 ± 4,9, z-score IMC: 4,4 ± 1,7; y 87 mujeres con IMC de 28,8 ± 4,6, z-score IMC: 3,8 ± 1,4) de entre los 4 y 18 años, a quienes se les practicó un test de tolerancia oral a la glucosa (TTOG) entre 1998 y 2004. Los resultados del TTOG se evaluaron siguiendo los criterios de la OMS y se calcularon parámetros de secreción y sensibilidad a la insulina (HOMA, QUICKI, área bajo la curva de glucemia y de insulina y el índice insulinogénico).
Resultados: La frecuencia de la intolerancia a la glucosa fue del 19,2 %. Sin embargo, ésta varía con la edad y el estadio puberal (prepuberal: 7,5 %, puberal: 39 % y pospuberal: 36 %) y con el IMC (z-score IMC entre +2 y +3: 8,7 %; entre +3 y +4: 21,5 % y mayor de +4: 25 %). Un paciente cumplía criterios de diabetes tipo 2.
Conclusiones: Los niños y adolescentes obesos de nuestro medio presentan una elevada incidencia de intolerancia a la glucosa que parece estar muy directamente relacionada con el grado de adiposidad.
