Sr. Editor:
Presentamos el caso de un lactante de 2 meses sin antecedentes patológicos con importante trabajo respiratorio de inicio progresivo. Está afebril y la presión arterial es de 90/40 mmHg, con una frecuencia cardíaca de 120 lat./min, y frecuencia respiratoria de 45 resp./min. No se observa en la exploración física estridor y la auscultación cardíaca y respiratoria es normal. En la radiografía de tórax no se aprecia congestión pulmonar.
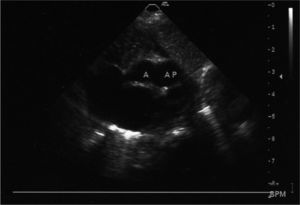
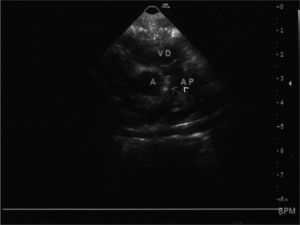
Se realizó ecocardiograma transtorácico: situs y concordancias adecuadas. Válvula pulmonar fina con buena movilidad y con regurgitación moderada. El tronco de la arteria pulmonar está dilatado con hipertensión pulmonar grave. Existe un defecto estructural en el septo aortopulmonar, antes de la división de las ramas pulmonares con comunicación anómala entre aorta ascendente y arteria pulmonar.
Con el diagnóstico de ventana aortopulmonar de tipo I se inició tratamiento con furosemida, digital y oxígeno. A las 48 h se realizó intervención quirúrgica con sellado mediante parche del defecto. El postoperatorio no tuvo complicaciones.
El defecto del septo aortopulmonar es una cardiopatía congénita rara. Según la clasificación de Richardson, se pueden diferenciar tres tipos. El tipo I o proximal es debido a la división inadecuada del tronco aortopulmonar; el tipo II es más distal e incluye el origen de la arteria pulmonar derecha, y el tipo III está constituido por un origen anómalo de la arteria pulmonar derecha que nace desde la aorta ascendente sin ninguna otra comunicación entre aorta y pulmonar.
Puede asociarse a otras cardiopatías como transposición de grandes vasos, tetralogía de Fallot, arco aórtico hipoplásico y atresia pulmonar.
Se debe corregir el defecto antes de los 6 meses de vida para evitar el desarrollo de enfermedad vascular pulmonar irreversible.