


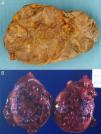









En este artículo se revisa y resume el estado actual del conocimiento de los 2 grandes grupos de tumores originados en la glándula suprarrenal: a) corticosuprarrenalomas, tumores derivados de la corteza de la glándula suprarrenal; y b) feocromocitomas y paragangliomas, tumores neuroendocrinos que tienen su origen en los paraganglios, formados por cúmulos ganglionares de células derivadas de la cresta neural, que se distribuyen simétricamente a lo largo del sistema nervioso autónomo, desde la pelvis a la base del cráneo, siguiendo el eje longitudinal del cuerpo (paragangliomas [PG]). Estos últimos (PG) pueden ser funcionantes y secretar catecolaminas que, al oxidarse con sales de cromo, adquieren un color marrón oscuro (tumores cromafines). Entre ellos, el término de feocromocitoma (FC) se reserva a los PG derivados de las células cromafines de la médula suprarrenal (PG intra-suprarrenales o de médula suprarrenal); mientras que, el término de PG hace referencia a los PG localizados fuera de la glándula suprarrenal, tanto simpáticos como parasimpáticos.
Se analizará el estado actual de las bases conceptuales, patogénicas, fundamentos genéticos y elementos diagnósticos (manifestaciones clínicas, parámetros bioquímicos y hormonales, técnicas de imagen y estudios moleculares) y terapéuticos (cirugía, tratamiento médico pre y postoperatorio, quimioterapia y radioterapia) de aplicación en la actualidad o en desarrollo.
This special article aims to summarise the current knowledge regarding the two groups of tumours with their origin in the adrenal gland: 1) adrenocortical tumours, derived from the cortex of the adrenal gland and 2) phaeochromocytomas and paragangliomas, neuroendocrine tumours derived from nodes of neural crest derived cells symmetrically distributed at both sides of the entire spine (paragangliomas [PG]). These PGs can be functioning tumors that secrete catecholamines, which confers their typical dark colour after staining with chromium salts (chromaffin tumors). Among these, the term phaeochromocytoma (PC) is restricted to those PGs derived from the chromaffin cells in the adrenal medulla (intra-adrenal PGs), whereas the term PG is used for those sympathetic or parasympathetic ones in an extra-adrenal location.
We analyse the state of the art of their pathogenic and genetic bases, as well as their clinical signs and symptoms, the tests currently available for performing their diagnosis (biochemical, hormonal, imaging and molecular studies) and management (surgery, pre- and post-surgical medical treatment), considering the current and developing strategies in chemo- and radiotherapy.
Existen 2 grandes grupos de tumores originados en la glándula suprarrenal:a) tumores corticosuprarrenales o corticosuprarrenalomas, tumores derivados de la corteza de la glándula suprarrenal; y b) feocromocitomas y paragangliomas, tumores neuroendocrinos que tienen su origen en los paraganglios, formados por cúmulos ganglionares de células derivadas de la cresta neural que se distribuyen simétricamente a lo largo del sistema nervioso autónomo, desde la pelvis a la base del cráneo, siguiendo el eje longitudinal del cuerpo (paragangliomas [PG]). Estos últimos (PG) pueden ser funcionantes y secretar catecolaminas que, al oxidarse con sales de cromo, adquieren un color marrón oscuro (tumores cromafines) (fig. 1). Entre ellos, el término de feocromocitoma (FC) se reserva a los PG derivados de las células cromafines de la médula suprarrenal (PG intra-suprarrenales o de médula suprarrenal); mientras que el término de PG hace referencia a los PG extra-suprarrenales, tanto simpáticos como parasimpáticos.
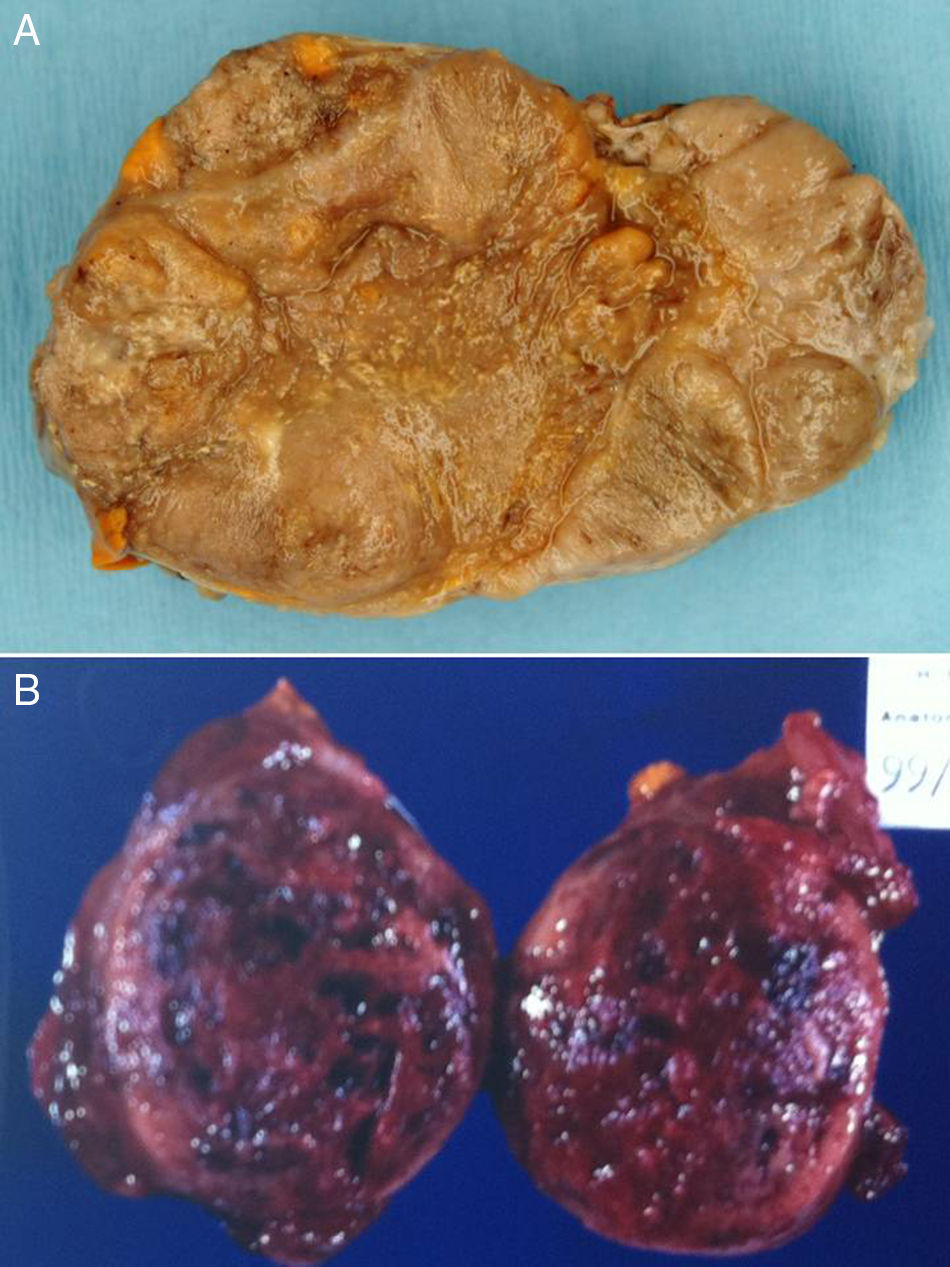
En 1948 Lawson Wilkins publicó 2 interesantes casos clínicos sobre un tumor suprarrenal feminizante que causaba ginecomastia en un varón de 5 años y sobre un tumor virilizante en una niña de 5años de edad1. Gran parte de las reflexiones entonces emitidas serían similares en la actualidad. Una revisión de 34 casos publicada en 19752 y otra más reciente del año 20013 demuestran el lento avance experimentado en el conocimiento del tratamiento de los tumores corticosuprarrenales en las últimas décadas.
Los feocromocitomas y los paragangliomas son igualmente raros en la infancia y benignos en la mayoría de los casos. El signo más frecuente al diagnóstico es la hipertensión arterial, asociada a episodios de cefaleas con palpitaciones y palidez. El estudio de los síndromes familiares de feocromocitomas ha resultado de gran importancia en la comprensión de los mecanismos patogénicos, tanto en casos familiares como esporádicos. En la actualidad se han implicado 10 genes diferentes en la patogénesis de estos tumores, sugiriéndose la posible participación de otros genes, particularmente en los casos esporádicos.
Tumores corticosuprarrenalesEpidemiologíaLos tumores derivados de las células de la corteza suprarrenal o tumores corticosuprarrenales (TCS) son raros en niños y adolescentes, de presentación heterogénea y patogenia aún entendida parcialmente. Se presentan siguiendo una tendencia bimodal, siendo más frecuentes en los primeros 5 años de la infancia y en la cuarta y quinta décadas en la vida adulta. En todos los rangos etarios son más frecuentes en el sexo femenino.
En los niños el tumor de origen suprarrenal más prevalente es el neuroblastoma, mientras que los TCS en su conjunto son extraordinariamente infrecuentes, por lo que prácticamente ningún centro endocrinológico u oncológico pediátrico atesora experiencia en su diagnóstico y seguimiento. A esta dificultad se suma la particularidad, con respecto a los casos de pacientes adultos, de que, independientemente de su benignidad o malignidad, los tumores suprarrenales secretores de esteroides sexuales pueden determinar un efecto deletéreo sobre el crecimiento del niño debido a una maduración sexual y somática precoces y patológicas.
Los datos del Surveillance, Epidemiology and End Results (SEER) procedentes del National Cancer Institute de EE. UU. estiman que solamente el 1,3% de los procesos de la infancia son carcinomas, siendo apenas un 0,2% TCS. Se considera que en EE. UU. se diagnostican anualmente 25 nuevos casos de TCS, con 12.400 casos de cáncer/año en menores de 20 años. No obstante, existen variaciones geográficas, siendo la máxima en el sudeste de Brasil (aproximadamente 15 veces superior: 3,4 casos por millón de niños menores de 14 años y por año). En 1990 se fundó el International Pediatric Adrenocortical Tumor Registry (IPACTR) en el St. Jude Children's Research Hospital de Memphis (EE. UU.) y el Hospital de Clínicas de Curitiba (Brasil), con el objetivo de obtener amplia experiencia al disponer de un mayor número de casos clínicos, muestras de laboratorio, tejidos y estudios moleculares, además de investigar nuevas vías terapéuticas (www.stjude.org/international-outreach). En 2002 el Children's Oncology Group (COG) creó el Comité de tumores raros, incluyendo un subcomité para el estudio de los TCS4–8.
EtiologíaLos mecanismos de desarrollo tumoral en la corteza suprarrenal se conocen parcialmente, fundamentándose nuestro conocimiento en el estudio de las alteraciones genéticas presentes en sus asociaciones patológicas y en los mecanismos de señalización hormonal de la corteza suprarrenal. La clasificación es compleja, basándose preferentemente en los estadios tumorales. Pueden ser benignos o malignos, funcionantes (secretores de hormonas) o no funcionantes5–8. Las bases genéticas se han desarrollado ampliamente en los últimos años. Las mutaciones en el gen supresor tumoral p53 (TP53) son las más comunes, tanto en los corticosuprarrenalomas aislados, como en aquellos asociados a otras entidades9, como es el síndrome de Li-Fraumeni. En el caso del complejo de Carney10,11 las mutaciones más frecuentes afectan al gen PRKAR1A12–14.
Alteraciones genéticas en los tumores corticosuprarrenalesLa aproximación a las bases moleculares de los TCS se ha realizado empleando múltiples técnicas de rastreo hologenómico, dirigidas a la búsqueda de fenómenos de pérdida o ganancia de determinadas regiones cromosómicas. Así, se ha demostrado mediante hibridación genómica comparativa (HGC) que existe una relación entre el tamaño tumoral y el número de ganancias o pérdidas de material cromosómico, sugiriendo que estas se acumulan conforme el tumor progresa. Las alteraciones cromosómicas más comunes observadas en niños con TCS son la ganancia de copias de la región cromosómica 9q34 y la deleción en homozigosis de la región 10p2115.
Entre los oncogenes implicados en estas alteraciones moleculares destacan RAS (adenomas productores de aldosterona), β-catenina, IGF-II y otros factores de crecimiento; y genes implicados en la supresión tumoral, de los que el TP5316 y el MEN1 son los principales exponentes. Las mutaciones activadoras de los primeros e inactivadoras de los segundos parecen implicadas en la génesis de los TCS, en particular, en determinadas asociaciones patológicas. Una reciente revisión analiza en detalle las bases genéticas en los pacientes con TCS17.
Asociaciones patológicas de los tumores corticosuprarrenalesSíndrome de Li-FraumeniEs un síndrome de cáncer hereditario muy heterogéneo. Clínicamente se caracteriza por un patrón de herencia autosómico dominante (AD) y la aparición, en edades tempranas, de múltiples tumores en varios miembros de un mismo grupo familiar. El tipo de tumores que pueden presentar los pacientes afectos de síndrome de Li-Fraumeni (SLF) es variado, siendo los más comunes los de tejidos blandos, osteosarcomas, mama, cerebro, TCS y leucemia18.
El 70% de los casos de SLF presentan mutaciones del gen supresor tumoral TP53 (Mendelian Inheritance in Man [MIM] 191170)9,19, localizado en 17p13.1 y que codifica la proteína p53. Al SLF secundario a mutaciones en el gen PT53 se le denomina SLF tipo 1 (MIM 151623), puesto que en otros subgrupos de pacientes afectos de SLF sin mutación en el gen PT53 se han hallado mutaciones en el gen CHEK2 (SLF tipo 2; MIM 609265; 22q11) y en un tercer locus en posición 1q23 (SLF tipo 3; MIM 609266).
Más del 90% de los niños menores de 4 años con TCS presentan una mutación del gen TP53. Por ello se ha sugerido que los TCS que aparecen en esta edad derivan de la zona fetal de la corteza suprarrenal, con características clínicas diferentes de los TCS del adolescente. La mutación germinal R337H de este gen, que afecta a los dominios de oligomerización de la proteína, es la más común en los pacientes de origen brasileño.
Neoplasia endocrina múltiple tipo 1 (MEN1, MIM 131100)De localización pancreática, hipofisaria y paratiroidea, preferentemente, se asocia hasta en el 36% de los casos con la presencia de nódulos en la corteza suprarrenal. La proteína codificada por el gen MEN1 es la menina. La asociación entre mutaciones germinales de MEN1 (11q13) y TCS se ha observado exclusivamente en pacientes adultos.
Síndrome de Beckwith-Wiedemann (MIM 130650)Es debido, de forma casi unívoca, a mutaciones en el locus del factor de crecimiento similar a la insulina número ii (IGF-II, 11p15.5), que determinan la pérdida de heterozigosidad y un defecto de impronta (sobreexpresión del alelo paterno) que ocasiona un exceso de expresión de IGF-II.
Tienen un mayor riesgo de padecer diversos tumores, tanto benignos como malignos, entre los que destacan el tumor de Wilms y, en segundo lugar, los TCS.
Complejo de CarneyEste síndrome de cáncer hereditario (patrón AD en el 50% de los casos)10,11 es secundario a mutaciones en 2 loci: PRKAR1A en 17q22-q24 (tipo 1, MIM 160980) y 2 genes fosfodiesterasa: PDE11A y PDE8B en 2p16 (tipo2, MIM 605244).
La principal manifestación endocrinológica del complejo de Carney es la enfermedad adrenocortical nodular pigmentada primaria, asociándose a múltiples tipos de tumores entre los que destacan los mixomas. Recientemente se han publicado 2 casos asociados a TCS20,21 Asimismo, se han hallado mutaciones somáticas y situaciones de pérdida de heterozigosidad en el gen PRKAR1A en TCS esporádicos12–14.
También se ha descrito un aumento de la incidencia de TCS en pacientes con hiperplasia suprarrenal congénita, neurofibromatosis tipo 1 o síndrome de McCune Albright.
Anatomía patológica de los tumores corticosuprarrenalesNo existe correlación unívoca entre el tamaño del tumor o sus características histopatológicas y su capacidad de producción hormonal, como no existe una característica patológica individual que determine la malignidad de un TCS. Hoy no se aconseja el empleo de la puntuación o score de Weiss, siendo la clasificación actual de los estadios tumorales (IPACTR) la más empleada para estimar el pronóstico del paciente4,5,7:
- •
Estadio I: tumor completamente extirpable,<100g y<200cm3 con niveles hormonales normales en el postoperatorio.
- •
Estadio II: tumor completamente extirpable, ≥ 100g y/o ≥ 200cm3 con niveles hormonales normales en el postoperatorio.
- •
Estadio III:
Tumor no extirpable.
Tumor diseminado.
Pacientes con estadio i o ii que no normalizan sus niveles hormonales en el postoperatorio.
Pacientes con afectación nodular linfoide retroperitoneal.
- •
Estadio IV:
Presencia de metástasis a distancia.
La diferenciación entre el adenoma y el carcinoma suele ser difícil. Ningún parámetro aislado, excepto la existencia de metástasis, permite discriminar entre tumores benignos y malignos. Únicamente la microscopia electrónica puede ser de ayuda, dado que los carcinomas suelen mostrar morfología y número anormal de mitocondrias.
Manifestaciones clínicasEn el niño existen 2 procesos evolutivos, el crecimiento y la maduración somática y sexual, que pueden verse afectados por la hipersecreción hormonal de esteroides sexuales, ya sean masculinos (tumores virilizantes), o femeninos (tumores feminizantes) por parte de los TCS. La presentación con síntomas derivados de hipersecreción de glucocorticoides (síndrome de Cushing) o mineralocorticoides (síndrome de Conn) son infrecuentes22.
La forma más común de presentación de estos tumores en los niños es la virilización aislada (50-84% según las series) secundaria a la existencia, en la mayoría de los casos, de un carcinoma. Como consecuencia el varón prepuberal muestra aparición brusca y de progresión rápida de vello sexual púbico y axilar, acné, aumento de tamaño y pigmentación del pene y el escroto, con frecuentes erecciones, aceleración de la velocidad de crecimiento y de la maduración esquelética. El tamaño testicular permanece estable o se incrementa discretamente23 (fig. 2).
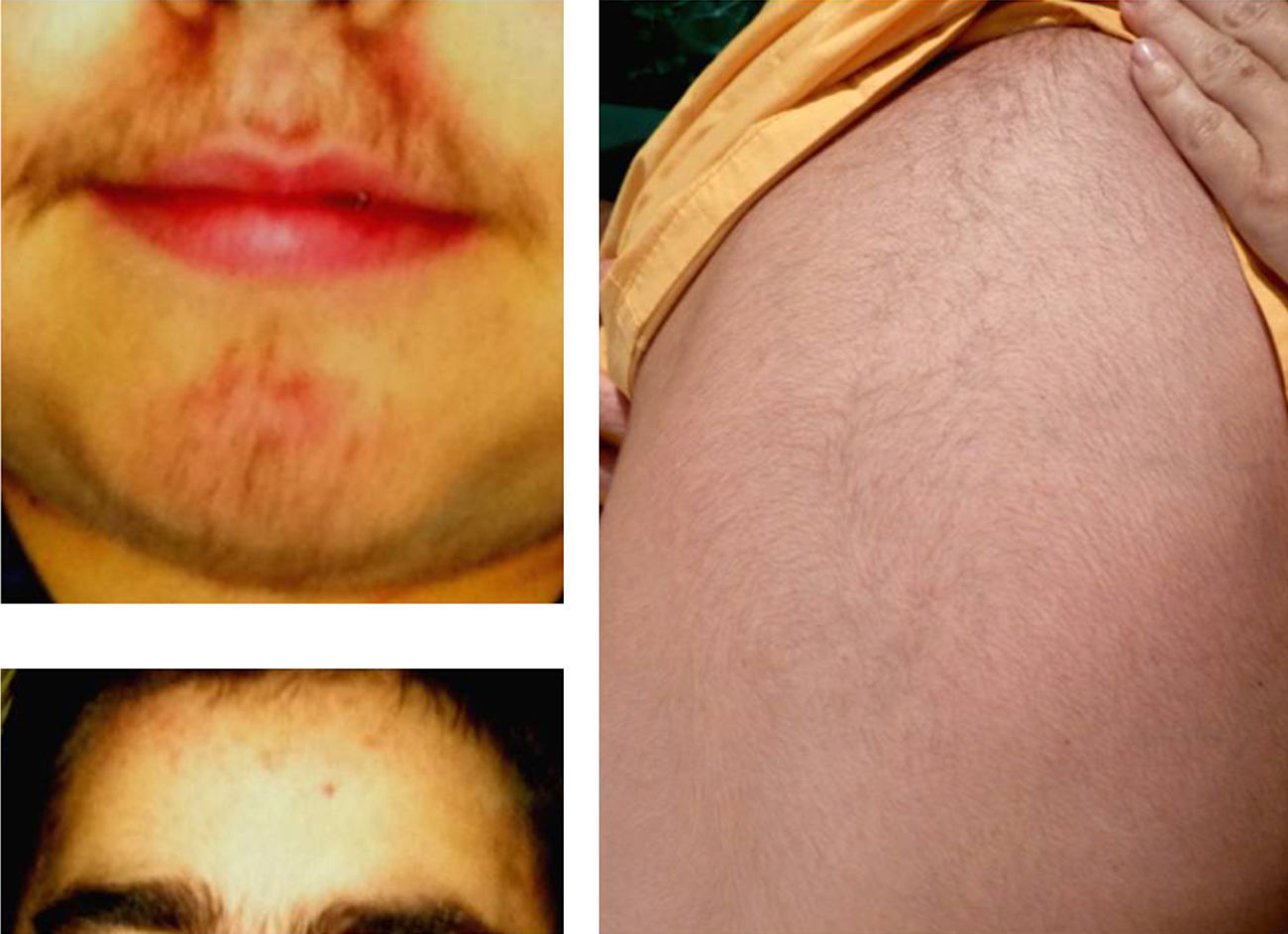
Las niñas afectas de TCS virilizantes muestran aparición del vello sexual púbico y axilar, acné y aceleración de la velocidad de crecimiento y de la maduración esquelética. Pueden presentar hirsutismo e hipertrofia y erectilidad del clítoris.
Los tumores feminizantes son mucho menos frecuentes (en torno al 7%, según algunas series), y el exceso de estrógenos ocasionado por los mismos determina, en el varón, la aparición brusca de ginecomastia, como signo más frecuente, acompañada de un incremento de la velocidad de crecimiento y de la maduración esquelética, mientras que no experimentan cambios en sus genitales externos ni aparición de vello sexual, salvo que coexista una hiperproducción de andrógenos asociada.
En la niña prepuberal generan un cuadro clínico completo de pseudopubertad precoz isosexual, con hipercrecimiento y progresión de la maduración esquelética, desarrollo mamario, cambios de pigmentación en los genitales externos y estrogenización del epitelio vaginal, apareciendo sangrado vaginal en aproximadamente el 50% de los casos. Si coexiste hipersecreción de andrógenos aparecerán los signos de la adrenarquia (vello púbico y axilar, cambio de secreciones apocrinas y acné)6.
DiagnósticoDebe comenzar con la realización de una historia clínica y examen físico detallado y orientado ante cualquier niño o niña con signos de virilización o feminización heterosexual o isosexual extemporánea, o ante el hallazgo de una masa abdominal.
- •
Evaluación analítica: determinación de las concentraciones plasmáticas de sodio y potasio circulantes, debido a la posibilidad de hipersecreción hormonal combinada, incluyendo cortisol, si bien la determinación de esteroides sexuales de producción suprarrenal y de sus metabolitos intermediarios constituirá la exploración fundamental. Ante TCS virilizantes se determinarán los niveles de: 17 cetoesteroides urinarios y/o dehidroepiandrosterona (DHEA), sulfato de DHEA (DHEA-S, de producción exclusiva adrenal), androstendiona y testosterona circulantes. Ante tumores feminizantes se determinarán niveles de estrógenos en plasma, incluyendo estradiol y estrona.
- •
Ante la sospecha clínica y/o analítica de la existencia de un TCS es preceptiva la realización de una prueba de imagen dirigida a la evaluación del área suprarrenal, con la precaución de que pueden existir restos de tejido suprarrenal en localización parahepática que podrían constituir la fuente de secreción hormonal.
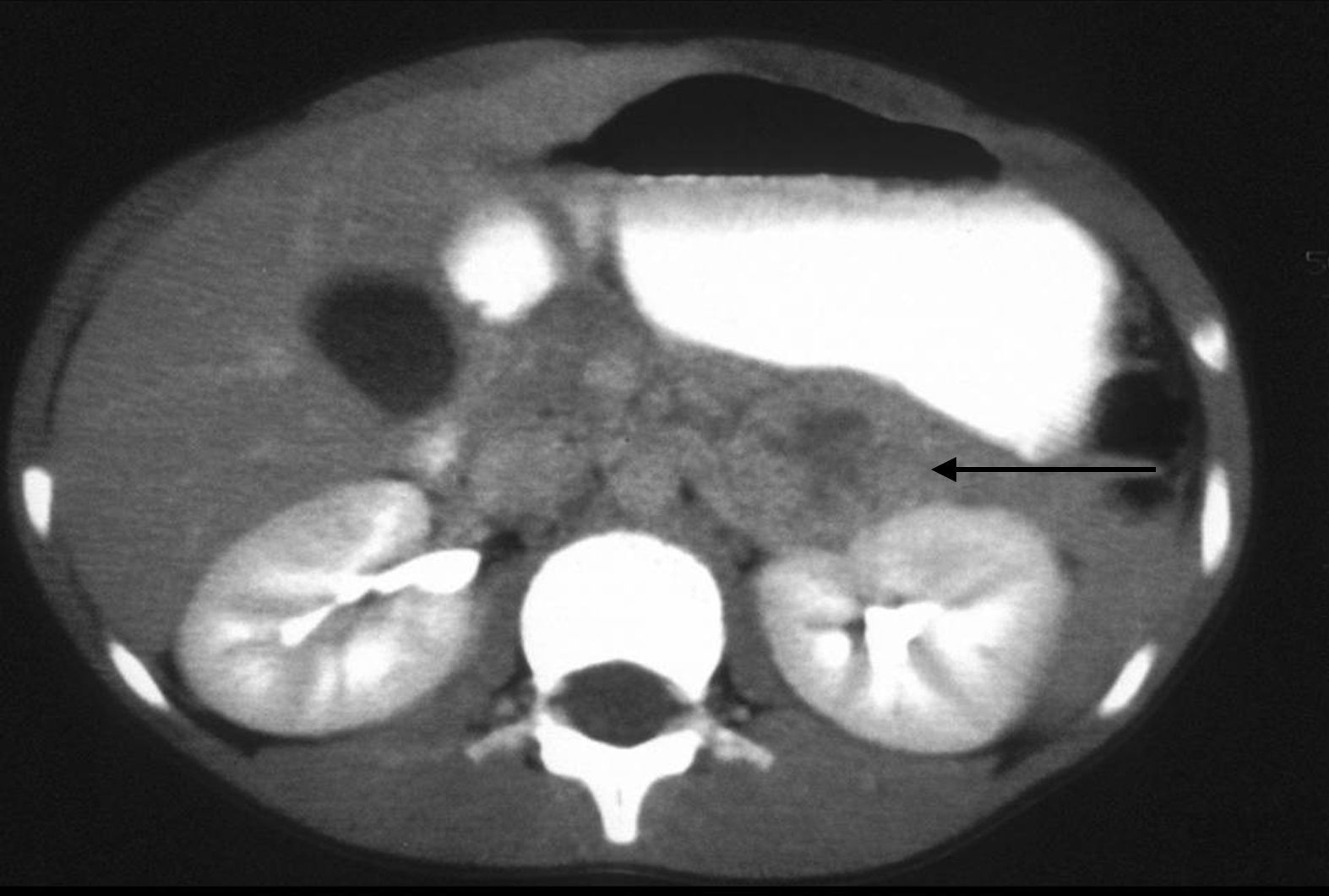
La ecografía, de fácil accesibilidad, sencilla realización y ausencia de radiación, tiene poca sensibilidad para la evaluación de las glándulas suprarrenales, más allá de los primeros meses de vida, por lo que no constituye la primera elección, si bien puede determinar la presencia de masas suprarrenales (fig. 3).
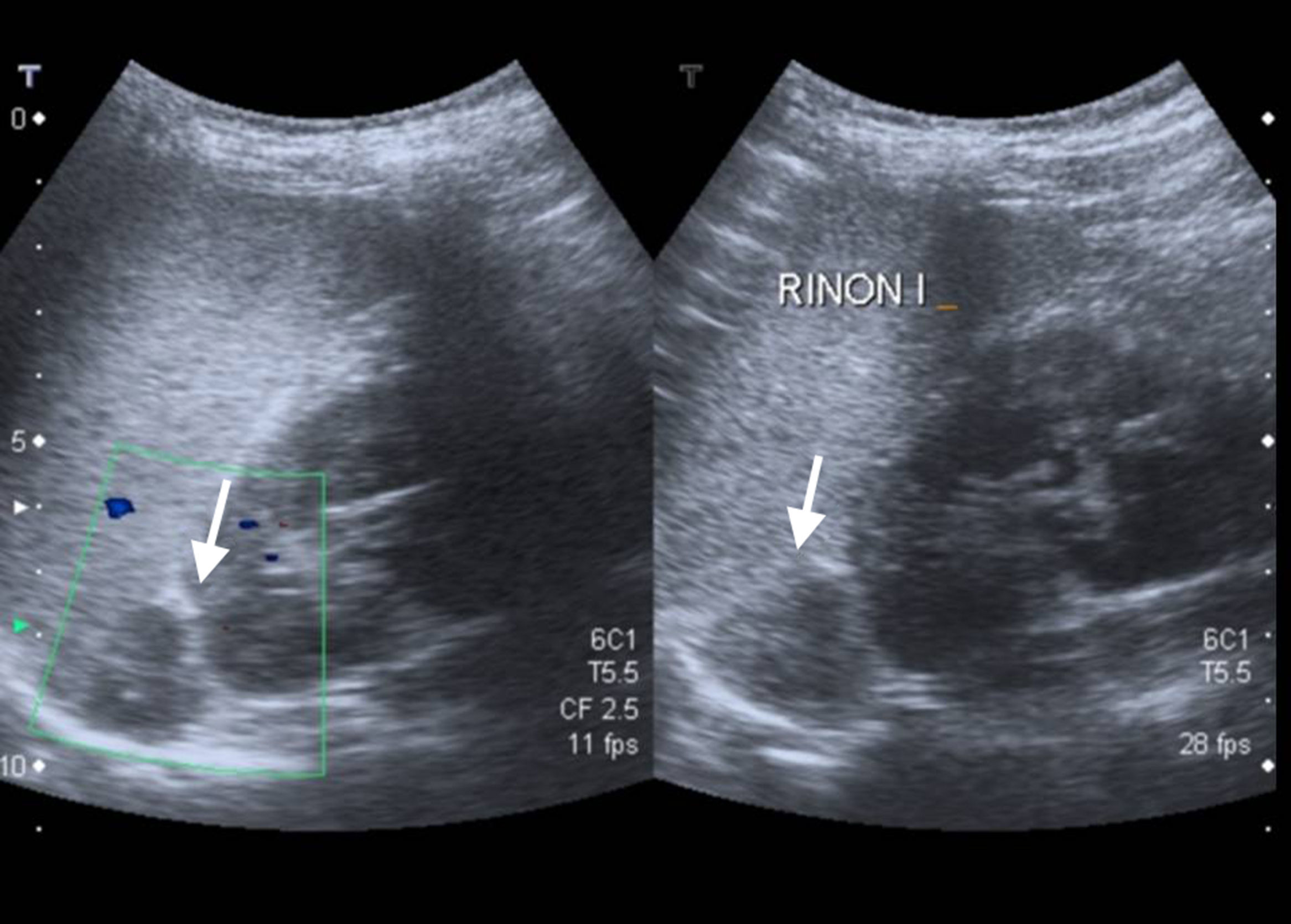
En cambio, se considera de elección para la evaluación de las glándulas suprarrenales la tomografía axial computarizada (TC) y la resonancia magnética (RM). Más recientemente se han empleado técnicas de imagen con radioisótopos como la tomografía por emisión de positrones (PET)24.
- •
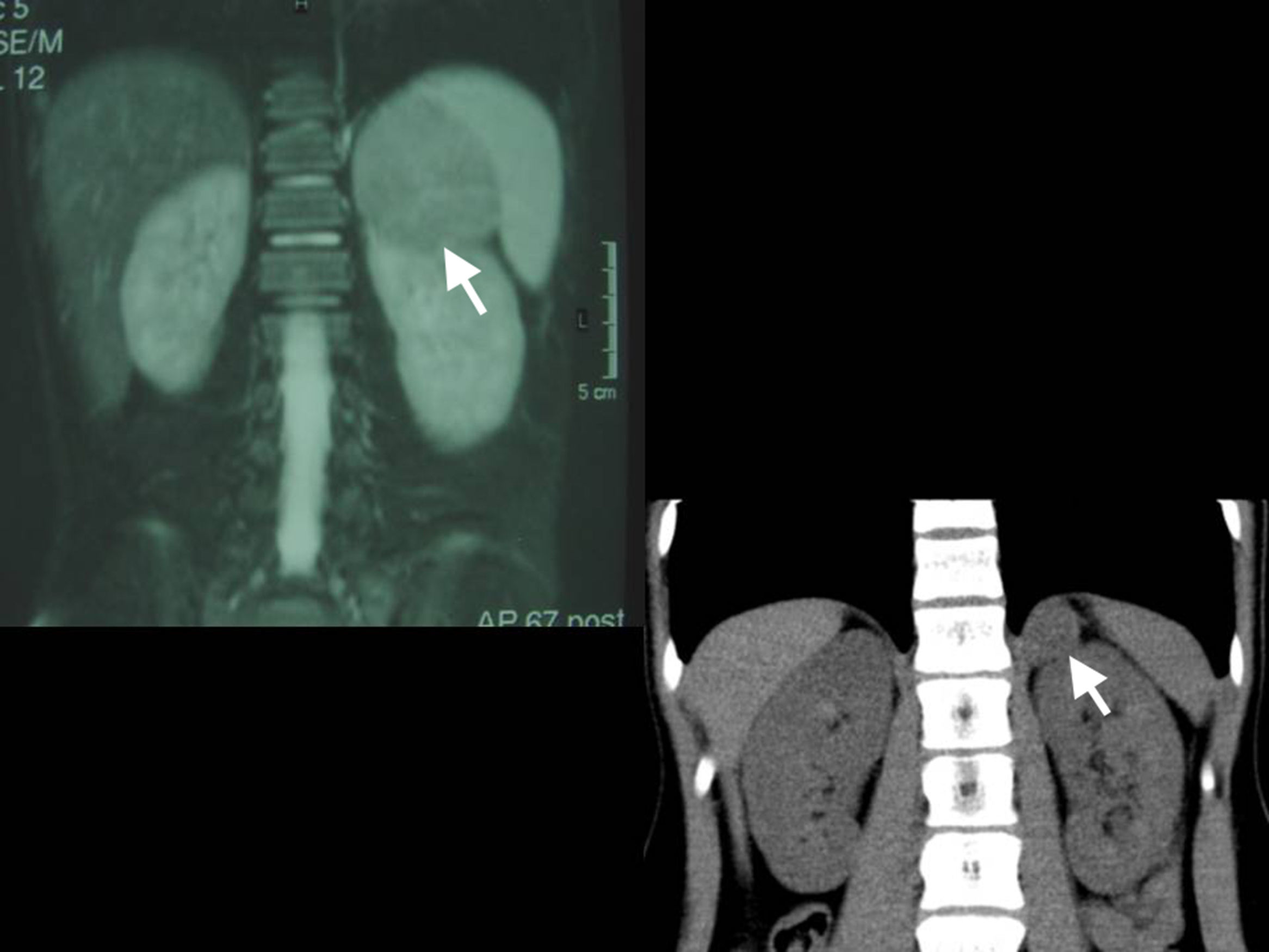
La TC es la técnica de elección para detectar y caracterizar la enfermedad suprarrenal. Debe ser ejecutada con colimación estrecha, tanto antes como después de la aplicación de un medio de contraste intravenoso, para obtener una adecuada resolución y medición de la densidad de las lesiones suprarrenales (fig. 4). El reciente advenimiento de la TC multidetectora permite la reconstrucción de secciones de 0,625mm y la conformación de imágenes multiplanares comparables a la RM. Su mayor limitación es el empleo de radiación ionizante, así como la necesidad de colaboración por parte del paciente, que puede conllevar la necesidad de sedación en niños pequeños. Asimismo, la TC es la técnica de elección para la detección de metástasis pulmonares o hepáticas24,25.
- •
Las ventajas de la RM, con o sin la administración de gadolinio, son la obtención de una imagen multiplanar y la ausencia de radiación ionizante, así como la mejor caracterización de una eventual invasión vascular (fig. 5). Por su parte, el empleo de radionúclidos ofrece información sobre actividad metabólica, basándose en la demostrada actividad metabólica de los TCS. El trazador más comúnmente empleado es la 18-fluorodesoxiglucosa (18-FDG), si bien se pueden emplear otros25 (fig. 6).
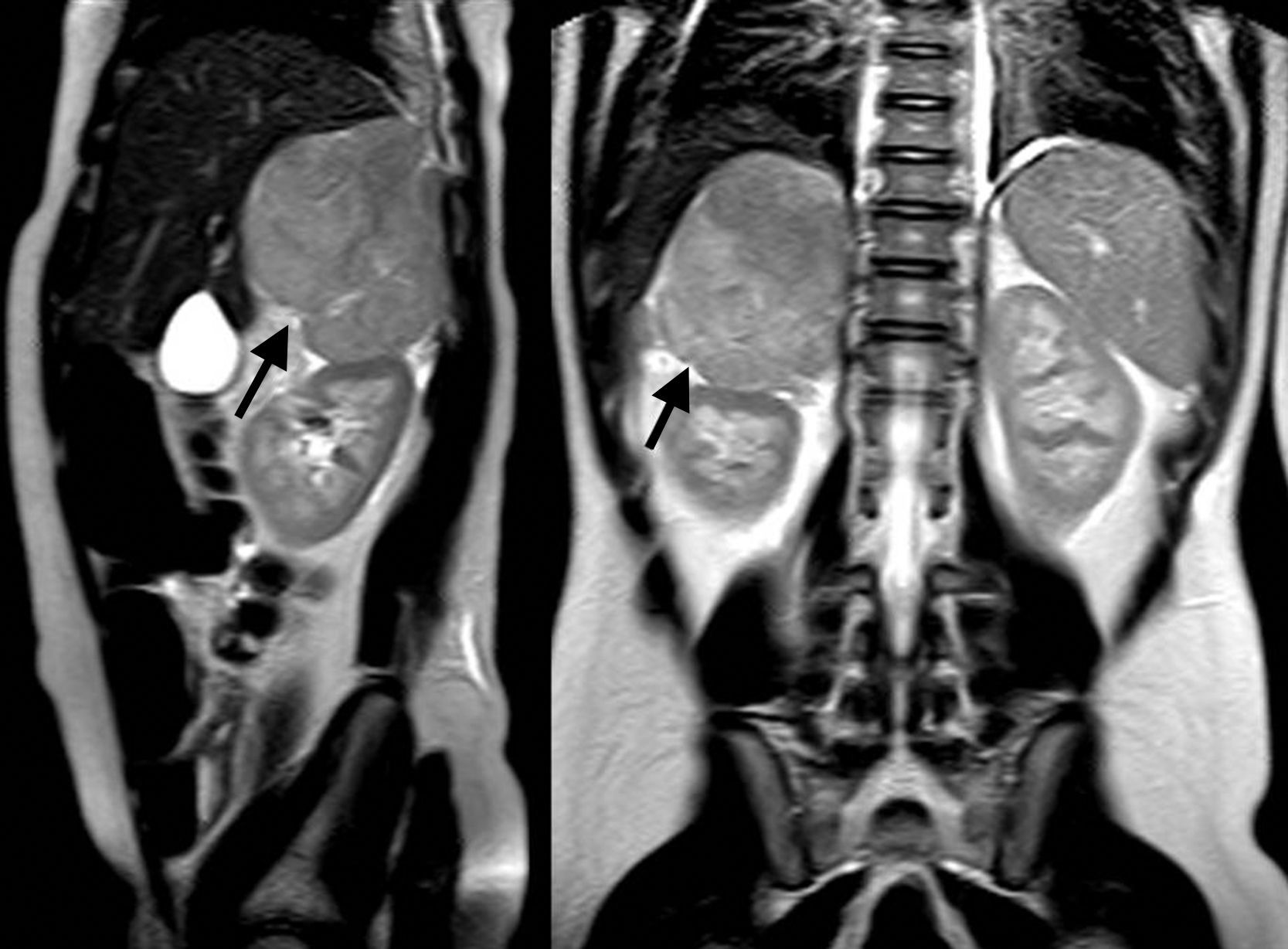
Las imágenes de PET se pueden combinar con las de TC (TC-PET), al igual que las de tomografía por emisión de fotón único (SPECT) (SPECT-TC), aunando información metabólica y definición anatómica25.
Un algoritmo de la secuencia diagnóstica ante la sospecha de un TCS se muestra en la figura 7.
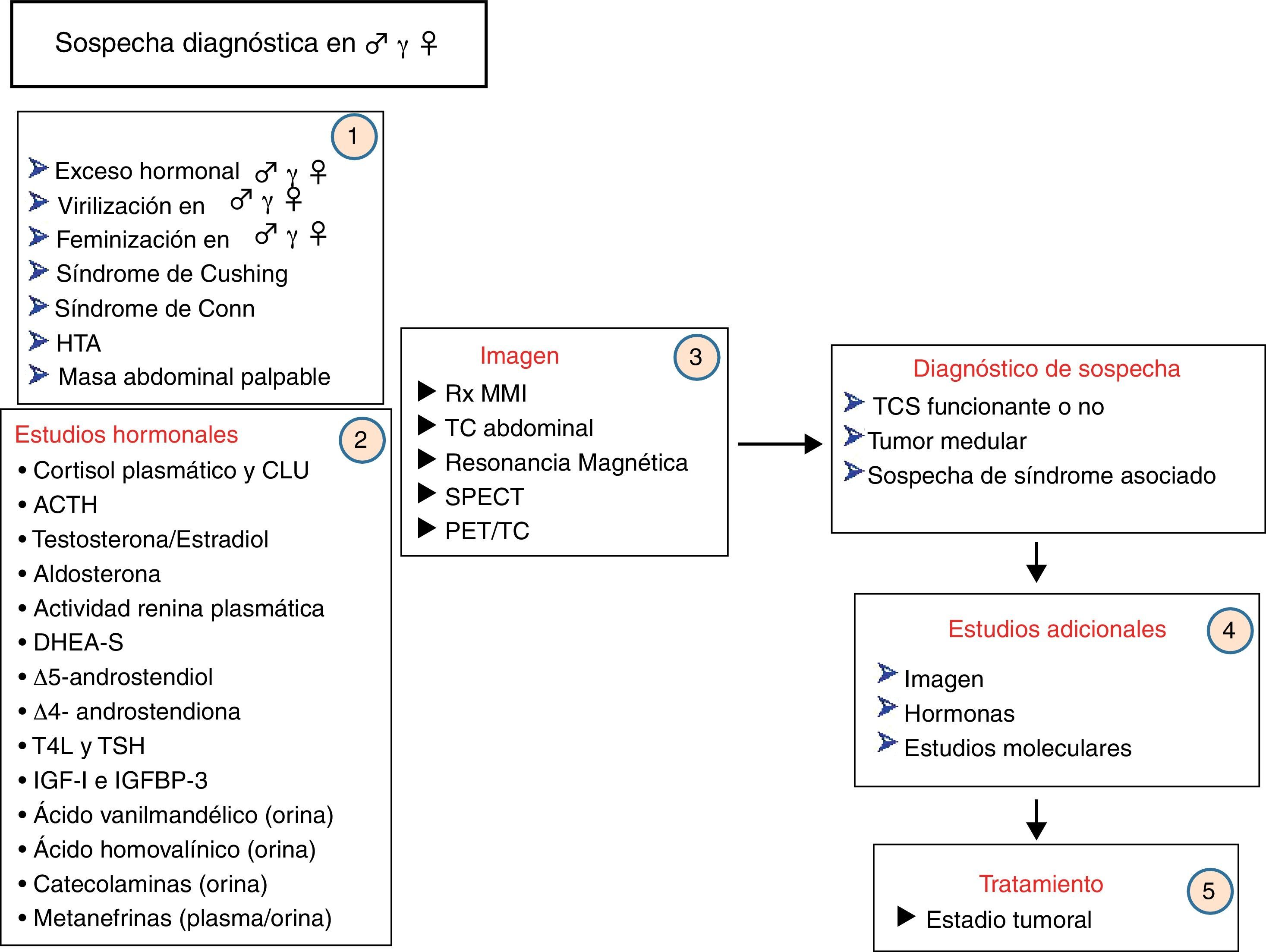
Algoritmo diagnóstico ante la sospecha de un tumor corticosuprarrenal.ACTH: hormona corticotropa; CLU: cortisol libre urinario en 24h; DHEA-S: sulfato de dehidroepiandrosterona; HTA: hipertensión arterial; IGF-I: factor de crecimiento similar a la insulina número 1; IGFBP-3: proteína transportadora de IGF-I número 3; PET: tomografía por emisión de positrones; RM: resonancia magnética; Rx MMI: radiografía de mano y muñeca izquierdas; SPECT: tomografía computarizada por emisión de fotón único; T4l: tiroxina libre; TC: tomografía axial computarizada; TCS: tumor corticosuprarrenal.; TSH: hormona tireotropa.
En la actualidad se estima con la nueva clasificación de los estadios tumorales antes indicada. Las conclusiones más importantes son las que siguen4:
- -
El pronóstico debe ser individualizado.
- -
La supervivencia en adenomas con resección completa es del 90% en estadios i y del 40% en estadios ii.
- -
Los carcinomas <200g en estadio i tienen un pronóstico favorable.
- -
Si el tumor se diagnostica en menores de 4 años la supervivencia es >70%, mientras que si se diagnostica entre los 4-12 años es del 30-40%.
- -
Los tumores que solo virilizan o son «no funcionantes» mejoran su pronóstico cuando se comparan con los que producen síndrome de Cushing.
- -
El desarrollo de las técnicas quirúrgicas mejorará el pronóstico.
- -
La terapia médica es paliativa, en espera de resultados de ensayos llevados a cabo en la actualidad.
Constituye la base esencial del tratamiento de los pacientes afectos de TCS en la infancia, hasta tal punto que la posibilidad de resección total del tumor, junto con el tamaño del mismo, son los 2 factores pronósticos prioritarios. Su indicación es aceptada de forma generalizada en los estadios i, iI y iii y, probablemente, en algunos casos en estadio iv.
Ante un TCS en estadio i se indica cirugía únicamente. Si el TCS se encuentra en estadio ii la cirugía debe acompañarse de la resección de nódulos linfoideos retroperitoneales. Si el TCS se encuentra en estadio iii o iv: quimioterapia (mitotane, asociación de cisplatino, etopósido y doxorrubicina) y cirugía más resección de nódulos linfoideos retroperitoneales4.
Mitotane y otros fármacos antineoplásicosEl mitotane (orto,para,dicloro-,difenil-,dicloroetano [O,p’DDD]) inhibe la síntesis hormonal por medio de la acción sobre las hormonas esteroidogénicas, es citotóxico frente a las células de la zona fascicular y reticular de la corteza suprarrenal e incrementa la producción de radicales libres. Fue aprobado para su empleo en los TCS por la FDA en 1970. Su uso en pacientes afectos de carcinomas de la corteza suprarrenal ha demostrado una reducción tumoral objetiva en el 25% de los casos, así como control de la secreción hormonal en la mayor parte de ellos.
Sus efectos secundarios son frecuentes y limitan el cumplimiento terapéutico (náuseas, vómitos, diarrea, dolor abdominal; y neurológicos: ataxia, vértigo, letargia y depresión). Estos efectos se aprecian a partir de concentraciones de 20mg/l.
Debido a su efecto adrenolítico, todos los pacientes tratados con mitotane deben recibir tratamiento sustitutivo con hidrocortisona y fludrocortisona por la posibilidad de insuficiencia suprarrenal que exacerba la intolerancia al mitotane.
Otros agentes antineoplásicos como la asociación de cisplatino, etopósido y doxorrubicina se están empleando en la actualidad4,7,8.
Radioterapia y otros tratamientosConsiderada tradicionalmente ineficaz en el tratamiento de los TCS, algunos de ellos pueden mostrar cierta sensibilidad a la misma. Constituye el tratamiento de elección en metástasis óseas y cerebrales, pudiendo ser empleada también en el tratamiento de recurrencias locales sintomáticas o en casos de resección incompleta4.
Está en desarrollo un protocolo de estudio colaborativo del tratamiento en niños afectos de TCS (ARAR0332) con el objetivo de evaluar la función del tratamiento exclusivamente quirúrgico en los TCS en estadio i, de la afectación de ganglios retroperitoneales en la recidiva local en TCS en estadio ii y el impacto de la quimioterapia con mitotane y otros en los TCS irresecables y/o metastásicos.
Las líneas experimentales de nuevas terapias frente a los TCS se dirigen hacia el desarrollo de agentes antineoplásicos alternativos a los existentes, inmunoterapia con células dendríticas, sustancias antiangiogénicas e inhibidores de la fosforilación de tirosinquinasas4.
El tratamiento de los pacientes con TCS requiere la colaboración de un equipo multidisciplinar, incluyendo: cirujano, oncólogo y endocrinólogo. Preoperatoriamente debe realizarse un estudio completo de las hormonas y precursores de producción suprarrenal, con el fin de identificar potenciales marcadores tumorales y suministrar al paciente una dosis de estrés de glucocorticoides previa a la resección tumoral. La terapia glucocorticoidea en situaciones de estrés debe mantenerse durante el tratamiento hasta que se confirme la normalidad de la función de la corteza suprarrenal4,7,8.
Una semana después de la intervención se debe repetir la evaluación hormonal, con el objetivo de confirmar la normalización o el drástico descenso de los niveles circulantes de los marcadores y hormonas elevadas inicialmente. Posteriormente, se recomienda efectuar reevaluaciones bimensuales durante el primer año, que pasarán a realizarse cada 4 meses en el segundo año y cada 6 meses con posterioridad. Asimismo, es necesaria la monitorización y el eventual tratamiento de efectos potenciales derivados de la hipersecreción hormonal, como la activación del eje gonadotropo por impregnación hormonal y la aparición de una pubertad precoz secundaria a esta7,8.
Existe controversia respecto a la reevaluación mediante técnicas de imagen. En efecto, mientras las recomendaciones del grupo internacional de consenso aconsejan realizarlas solamente en presencia de hallazgos hormonales.
Debido a su escasa frecuencia los TCS en la infancia constituyen un reto diagnóstico y terapéutico en el que el conocimiento y los recursos disponibles son muy limitados. La creación de grupos colaborativos de estudio y de un registro internacional supone un gran avance a este respecto, con el fin de agrupar la experiencia colectiva para poder ofrecer una mejor asistencia a nuestros pacientes que permita mejorar las tasas de supervivencia actuales.
Feocromocitomas y paragangliomasEpidemiologíaSon tumores raros, con una incidencia de 0,3 casos/106 habitantes/año; (≈ 10-20% se diagnostican en el niño). En este periodo la edad media de aparición es de 11 años, con ligero predominio en varones, especialmente en los menores de 10 años, y en la gran mayoría de los casos se trata de tumores benignos (85-90%) y de localización suprarrenal (FC)26,27. Sin embargo, pueden ser malignos en un 10% de los casos y es esencial valorar si la localización es exclusivamente suprarrenal o múltiple (extra-suprarrenal) y si existen antecedentes familiares de FC28.
EtiologíaLos FC/PG pueden presentarse de forma esporádica o en el contexto de síndromes tumorales familiares. Las formas familiares o sindrómicas son, en su mayoría, debidas a mutaciones germinales en genes que predisponen a su desarrollo y que se heredan con un patrón de herencia AD y penetrancia incompleta. Existen 10 genes implicados en la patogénesis de estos tumores29–33: VHL, RET, NF1, SDHA, SDHAF2, SDHB, SDHC, SDHD,TMEM127 y MAX (tabla 1), habiéndose descrito este último en el Centro Nacional de Investigaciones Oncológicas (CNIO) de Madrid, por el grupo de la Dra. Robledo34.
Formas familiares o sindrómicas de feocromocitoma/paraganglioma
| Gen (localización) y año de identificación | Síndrome asociado | Edad media (años) presentación del FC/PG (rango) | Porcentaje de todos los FC/PG (%) | Porcentaje mutaciones de novo (%) | Penetrancia de FC/PG (%) | Frecuencia de los tipos de tumor (%) | Malignidad (%) | Otras características clínicas |
| VHL (3p25-26) 1993 | Enfermedad von Hippel-Lindau | 28 (5-67) | 9,0 | 20 | 10-26 | FC: 90,3PG (s/ps):18,6 (5,9/8,8) | <5 | Hemangioblastomas de SNC y retinaQuistes renales y tumor renal de células clarasQuistes, tumores y cistoadenomas pancreáticosCistoadenoma del epidídimo y del ligamento redondoTumores del saco endolinfático (oído interno) |
| RET (10q11-21) 1993 | MEN2 | 35,6 (4-73) | 5,3 | MEN 2A: 5 MEN 2B: 50 | 50 | FC: 100%PG: excepcional | <5 | MEN 2A: FC, CMT y adenoma/hiperplasia de paratiroidesMEN 2B: FC, CMT, ganglioneuromatosis mucosa e intestinal y hábito marfanoide |
| SDHD (11q23.1) 2000 | PGL1 | 35 (10-96) | 7,1 | ¿? | 86 (alelo pat.)0 (alelo mat.) | FC: 23,9PG (s/ps): 91,5 (22/84) | <5 | Asociado a mutaciones en el alelo paternoPuede asociarse a síndrome Carney-Stratakis, caracterizado por la díada de PGL/FC (100%/9%) habitualmente múltiples y tumor estromal gastrointestinal (TEG) |
| SDH5/SDHAF2 (11q13.1) 2009 | PGL2 | 32,2 (25-59) | ≈ 0 | ¿? | ≈ 100 (alelo pat.)≈ 0 (alelo mat.) | FC: no se ha descritoPG (s/ps): 100 (0/100) | 0 | PG de la cabeza y el cuello multifocales que solo se asocian a mutaciones en el alelo paterno (solo se ha descrito un caso de transmisión materna) |
| SDHC (1q21) 2000 | PGL3 | 42,7 (13-73) | 0,5 | ¿? | ¿? | FC: excepcionalPG (s/ps): 100 (7/93) | ≈ 0 | Puede asociarse a síndrome de Carney-StratakisSolo se ha descrito un paciente con un PG maligno (del cuerpo carotídeo) |
| SDHB (1p35-p36.1) 2001 | PGL4 | 32,7 (6-77) | 5,5 | ¿? | 77% | • FC: 25,2• PG (s/ps): 77,5 (71/24) | 30,7 | Puede asociarse a síndrome de Carney-StratakisPuede asociar carcinoma renal y tiroideo |
| SDHA (5p15.33) 2010 | Sin síndrome asociado (¿PGL5?) | 40 (27-55) | ≈ 0 | ¿? | ¿? | FC: 16,7PG (s/ps): 83,3 (50/30,3) | 0-14,3 | • Mutaciones germinales en SDHA se asocian a enfermedades neurodegenerativas (síndrome de Leigh.) y se han descrito asociadas a TEG sin PGL |
| NF1 (17q11.2) 1990 | Neurofibromatosis tipo 1 | 41,6 (1-74) | 2,9 | 50% | 0,1-5,7 | FC: 95,3PG (s/ps): 6,1 (6,1/0) | 9,3 | Manchas café con leche, pecas axilares e inguinales, nódulos de Lish en el iris, gliomas en SNC y nervios ópticos, escoliosis, neurofibromas |
| TMEM127 (2q11.2) 2010 | Sin síndrome asociado | 42,8 (21-72) | <2 | ¿? | ¿? | FC: 95,7PG (s/ps): 8,7 (4,3/4,3) | 4,3 | Actúa como un gen supresor tumoralProbable transmisión a partir solo del alelo paterno, de forma similar a PGL1 y PGL2 |
| MAX (14q.23) 2011 | Sin síndrome asociado | 34 (15-58) | ≈ 1 | ¿? | ¿? | FC: 100PG (s/ps): 0 | 25 | Actúa como un gen supresor tumoralAlto riesgo de malignidad |
| ¿? 1999 | Tríada de Carney | 27,5 (12-48) | ≈ 0 | ¿? | No aplicable | FC: 16,2PG (s/ps): 15,8 (s/ps: ¿?) | 10,8 | Asociación de: PG, TEG y condromas pulmonaresPuede asociar otros tumores (leiomiomas esofágicos y adenomas adrenocorticales). Etiopatogenia no aclaradaAlrededor del 45% se presentan como PG/FC |
CMT: carcinoma medular de tiroides; FC: feocromocitoma; mat.: materno; pat: paterno; PG: paraganglioma; PGL: síndrome de paraganglioma familiar; ps: parasimpático; s: simpático; SNC: sistema nervioso central.
Tradicionalmente se consideraba que un 10% de estos tumores eran formas familiares o sindrómicas. Hoy se asume que el porcentaje es del 40-45%. Se deben en su mayoría a mutaciones germinales en alguno de los 10 genes conocidos que confieren susceptibilidad para el desarrollo de FC/PG (tabla 1) y, en ellas, es más frecuente el desarrollo de tumores múltiples o bilaterales (sincrónicos o metacrónicos), extradrenales y de aparición precoz29–34.
Las formas familiares más comunes en el niño se asocian a: enfermedad de von Hippel-Lindau (VHL), síndrome de neoplasia endocrina múltiple (MEN) tipo 2 y síndromes del paraganglioma familiar (SPGF). Con menos frecuencia: neurofibromatosis tipo 1 (NF1), tríada de Carney y mutaciones en TMEM127 y MAX. Excepcionalmente se han asociado a: MEN1, síndromes (Sturge-Weber, Pringle-Bourneville) y mutaciones en otros genes (KIF1B y EGLN1).
Enfermedad de VHLAunque es una entidad infrecuente (1:36.000), las mutaciones germinales en el gen supresor tumoral VHL son la principal causa genética de tumores cromafines en el niño. Suponen el 75% de las mutaciones observadas en FC/PG de aparición aparentemente esporádica antes de los 20 años. El 20-30% de los pacientes con VHL desarrollan FC/PG en su evolución y un 30-55% de los casos como primera manifestación de la enfermedad. En su mayoría son tumores suprarrenales (FC), benignos, noradrenérgicos (secretores de noradrenalina) y bilaterales (40-50%).
MEN 2Enfermedad infrecuente (prevalencia ≈ 1:40.000 individuos) causada por mutaciones germinales con ganancia de función en el protooncogén RET. Se distinguen 3 subtipos: MEN 2A (55%), MEN 2B (5-10%) y carcinoma medular de tiroides (CMT) familiar (35-40%). El 50% de los pacientes con MEN 2A y 2B desarrollan FC a lo largo de la vida, a menudo bilaterales (sincrónicos o metacrónicos) y adrenérgicos (secretores de adrenalina y noradrenalina), pero rara vez malignos. Determinadas mutaciones en el gen RET (codón 630, 634 y 883, entre otras) parecen conllevar un mayor riesgo de FC. Excepcionalmente se ha descrito el desarrollo de PG simpáticos o parasimpáticos. En más del 75% de los casos el CMT precede a la aparición del FC.
SPGFSe denominan habitualmente como SPGF de tipo 1 a tipo 5 (PGL1 a PGL5) y son debidos a mutaciones en los genes que codifican para las 4 subunidades (SDHA, SDHB, SDHC, SDHD) de la enzima succinato deshidrogenasa (SDH) o para una proteína, recientemente descubierta (SDHAF2), que interviene en el ensamblaje de la SDH a la membrana interna de la mitocondria. La SDH forma parte de la cadena de transporte de electrones (complejo ii) y del ciclo de Krebs. Todos estos genes parecen funcionar como genes supresores tumorales. El patrón de herencia es AD con penetrancia variable; si bien, PGL1 y PGL2 parecen estar improntados y solo se hereda la enfermedad cuando el alelo afectado es el paterno. En conjunto, los SPGF representan alrededor del 13% de los FC/PG, con una prevalencia estimada de 1:20.000-50.000 individuos, la mayoría PGL1 y PGL4, y cuando secretan catecolaminas su patrón suele ser noradrenérgico.
- •
PGL1 (SDHD). PG multifocales de la cabeza y el cuello y, menos frecuentemente, FC y PG simpáticos.
- •
PGL2 (SDHAF2). Mutaciones en SDHAF2 se han descrito en solo 2 familias y con la misma mutación. Todos los tumores descritos son PG parasimpáticos benignos y multifocales de la cabeza y el cuello.
- •
PGL3 (SDHC). PG de la cabeza y el cuello, habitualmente solitarios y benignos, aunque se han descrito algunos casos aislados de FC y PG simpáticos.
- •
PGL4 (SDHB). PG simpáticos con alto riesgo de malignidad, agresividad y precocidad, habitualmente de localización abdominal y funcionantes.
- •
PGL5 (SDHA). Las mutaciones en SDHA se han descrito en un número muy limitado de pacientes no relacionados y no se ha descrito un síndrome específico asociado. Parecen predisponer a PG simpáticos y parasimpáticos aislados y preferentemente benignos.
La etiopatogenia de los FCS/PG esporádicos es desconocida, aunque el hallazgo de alteraciones genéticas en el tejido tumoral es relativamente frecuente (mutaciones somáticas en el gen supresor tumoral p53 o pérdidas de heterozigosidad cromosómica). Por el contrario, el hallazgo de mutaciones somáticas de los genes implicados en las formas familiares es escasa. Un 25-30% de los FC/PG aparentemente esporádicos se debería a mutaciones germinales en alguno de los genes de susceptibilidad, bien por mutaciones de novo bien por ausencia o desconocimiento de antecedentes familiares. Este porcentaje se incrementa a medida que la edad de presentación es menor (<5% en mayores de 40 años, alrededor del 60% en menores de 20 años y alrededor del 70% en menores de 10 años). Por este motivo los casos esporádicos de FC deben, asimismo, estudiarse molecularmente35,36.
Anatomía patológicaSolo un 10% de los FC/PG en la edad pediátrica son malignos. No existen imágenes histológicas o inmunohistoquímicas que indiquen el potencial metastásico de este tipo de tumores. Por consiguiente, la biopsia preoperatoria no solo no está indicada, sino que es potencialmente peligrosa. La malignidad se establece por la presencia de metástasis a distancia.
Manifestaciones clínicasLa mayoría de los FC y PG simpáticos sintetizan y secretan catecolaminas (adrenalina, noradrenalina y, excepcionalmente, dopamina) que son responsables de sus principales manifestaciones clínicas37. Por el contrario, solo el 5-20% de los PG parasimpáticos son capaces de producir cantidades significativas de catecolaminas, por lo que la mayoría suelen ser asintomáticos o manifestarse como una pequeña masa creciente indolora, que puede originar síntomas dependientes de su localización (pérdida de audición, acúfenos, tos, disfagia o ronquera, entre otros).
En la infancia la mayoría de los tumores son funcionantes y la sintomatología depende de la secreción de catecolaminas, pero también de la sensibilidad individual de cada paciente. Las manifestaciones clínicas pueden ser muy variadas (tabla 2), pero el síntoma más constante es la hipertensión arterial (los FC/PG representan el 1-2% de los casos de hipertensión arterial secundaria en los niños). La mayoría de los pacientes se presentan con la clásica tríada asociada a las crisis de hipertensión arterial: cefalea, sudoración profusa y palpitaciones37,38.
Manifestaciones clínicas y complicaciones de los feocromocitomas/paragangliomas funcionantes
| Manifestaciones clínicas | Complicaciones cardiovasculares |
| Hipertensión arterial con/sin crisis paroxísticas | Infarto miocárdico |
| Cefalea | Miocarditis |
| Sudoración profusa | Infarto cerebral |
| Taquicardia y palpitaciones | Infarto renal |
| Visión borrosa | Espasmo vascular periférico (Raynaud, gangrena) |
| Palidez cutánea y sudoración acra | Edema pulmonar |
| Náuseas y vómitos | |
| Temblor | Complicaciones metabólicas |
| Crisis de ansiedad | Acidosis láctica |
| Malestar general | Hiperglucemia e intolerancia a la glucosa |
| Dolor epigástrico o precordial | Hipercalcemia |
| Diarrea o estreñimiento | Elevación de hematocrito |
| Pérdida de peso o falta de ganancia de peso | |
| Intolerancia al calor o febrícula | |
| Convulsiones | |
| Síncope |
En los niños, al contrario que en los adultos, aunque con episodios de exacerbación, la hipertensión arterial suele ser mantenida. Algunos síntomas son, también, más frecuentes en niños que en adultos: sudoración, alteraciones visuales, náuseas, vómitos y pérdida de peso. Un pequeño porcentaje pueden ser asintomáticos y diagnosticarse casualmente en pruebas de imagen realizadas por otro motivo (incidentalomas) o, en las formas familiares, como consecuencia de un cribado familiar a partir de un caso índice. Dado el origen neuroendocrino de estos tumores pueden ocasionalmente cosecretar otras hormonas y producir síndromes paraneoplásicos de secreción ectópica: síndrome de Cushing (CRH o ACTH), gigantismo hipofisario (GHRH), hipercalcemia (PrPTH) o diarrea secretora (PIV), entre otros.
DiagnósticoBioquímicoHa mejorado en los últimos años debido a los avances en los análisis utilizados para detectar y cuantificar los niveles de catecolaminas o sus metabolitos en sangre u orina. En la actualidad la prueba de elección es la medición de metanefrinas fraccionadas (metanefrina y normetanefrina) en plasma o, si no es posible, en orina de 24h. Una elevación de los niveles de metanefrinas 4 veces por encima del rango de referencia (deben ser adecuados a la edad del paciente, especialmente en niños) se asocia a una probabilidad prácticamente del 100% de que se trate de un tumor secretor de catecolaminas38,39.
En raras ocasiones el tumor secreta preferentemente dopamina; en estos casos deben determinarse además sus metabolitos: ácido homovanílico y metoxitiramina. También deben determinarse los niveles séricos de cromogranina A, una proteína presente en la matriz soluble de los gránulos cromafines, que sirve de excelente marcador tumoral y que ha demostrado su utilidad en el diagnóstico y seguimiento de estos y otros tumores de estirpe neuroendocrina.
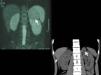
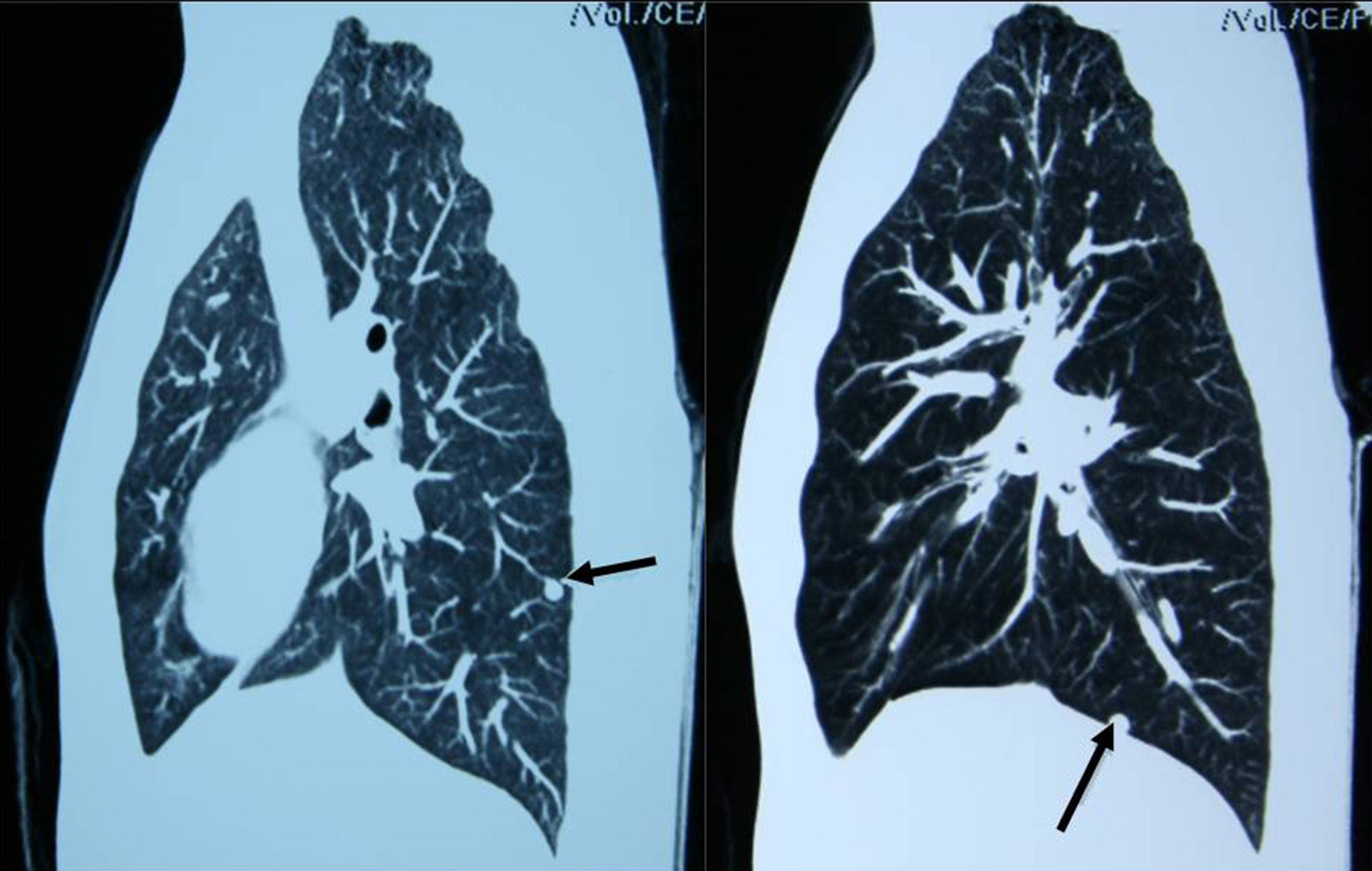
De localizaciónEstablecida la evidencia bioquímica de un FC/PG, el tumor debe ser localizado40. Las pruebas de primera elección son la TC y la RM, inicialmente de abdomen y pelvis y, si el estudio es negativo, de tórax y cuello. Ambas pruebas tienen una excelente sensibilidad diagnóstica, pero su especificidad es baja (figs. 8 y 9). La RM presenta algunas ventajas (mayor sensibilidad en PG, no requiere contraste y ausencia de radiación) que la hacen más indicada que la TC en niños y gestantes.
Para mejorar la especificidad diagnóstica se utilizan las imágenes funcionales. La escintigrafía corporal total con metaiodobencilguanidina (MIBG) marcada con 123I tiene una alta sensibilidad y especificidad, ya que puede confirmar la naturaleza secretora del tumor y localizar tumores no detectados por la TC/RM. Dado que la escintigrafía con [123I]-MIBG no posee una sensibilidad del 100%, especialmente en el caso de tumores malignos o metástasis, se han utilizado otros radiotrazadores, como el octreótido marcado con indio o iodo radioactivos (se fijaría al receptor de sosmatostatina) y modalidades de imagen nuclear (PET). La PET es una modalidad fisiológica de escintigrafía, con menor exposición a radiación y mayor rapidez en la obtención de imágenes. La PET puede utilizarse con [18F]-fluorodesoxiglucosa (FDG) o con trazadores más específicos para tejido cromafín ([18F]-DOPA o 6-[18F]-fluordopamina) (fig. 10).
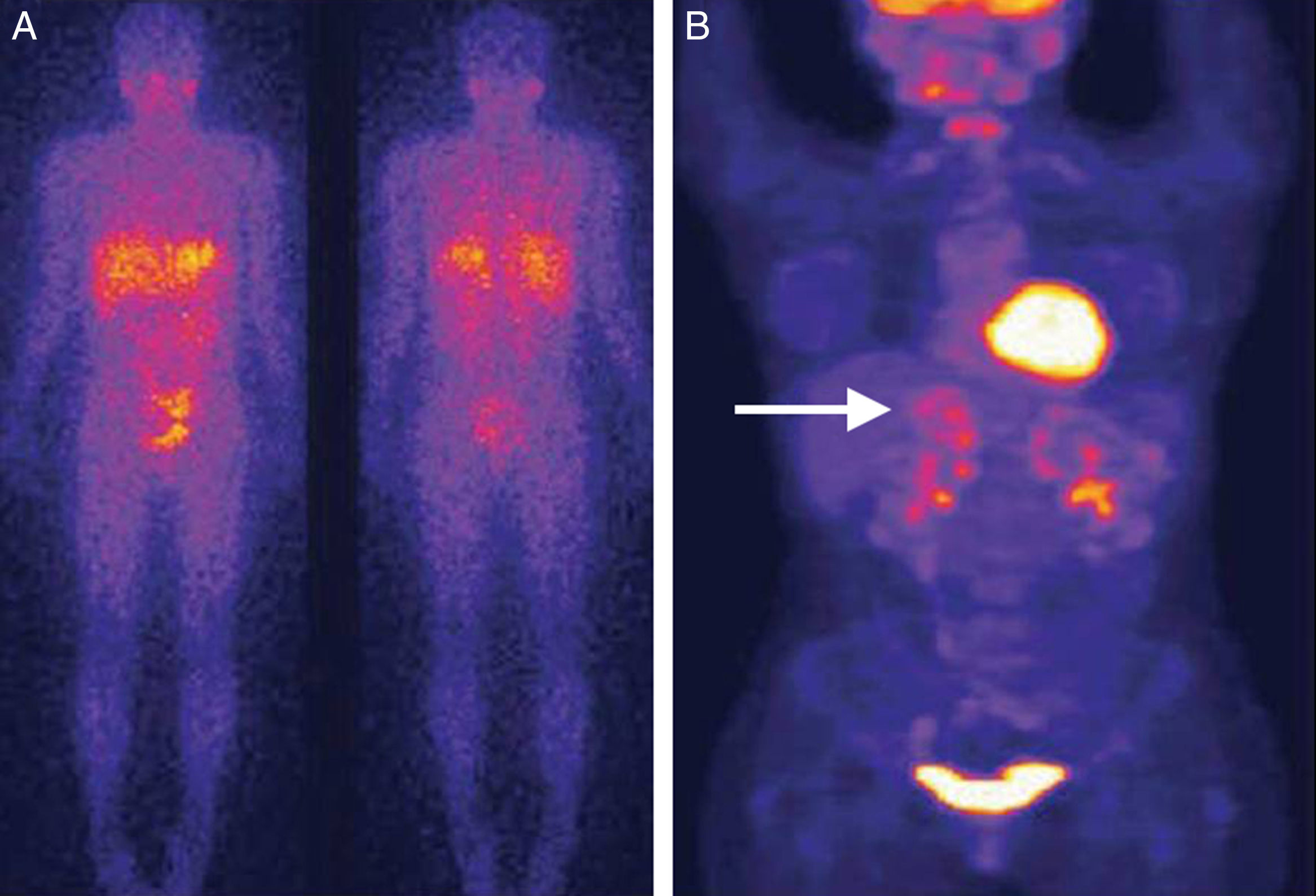
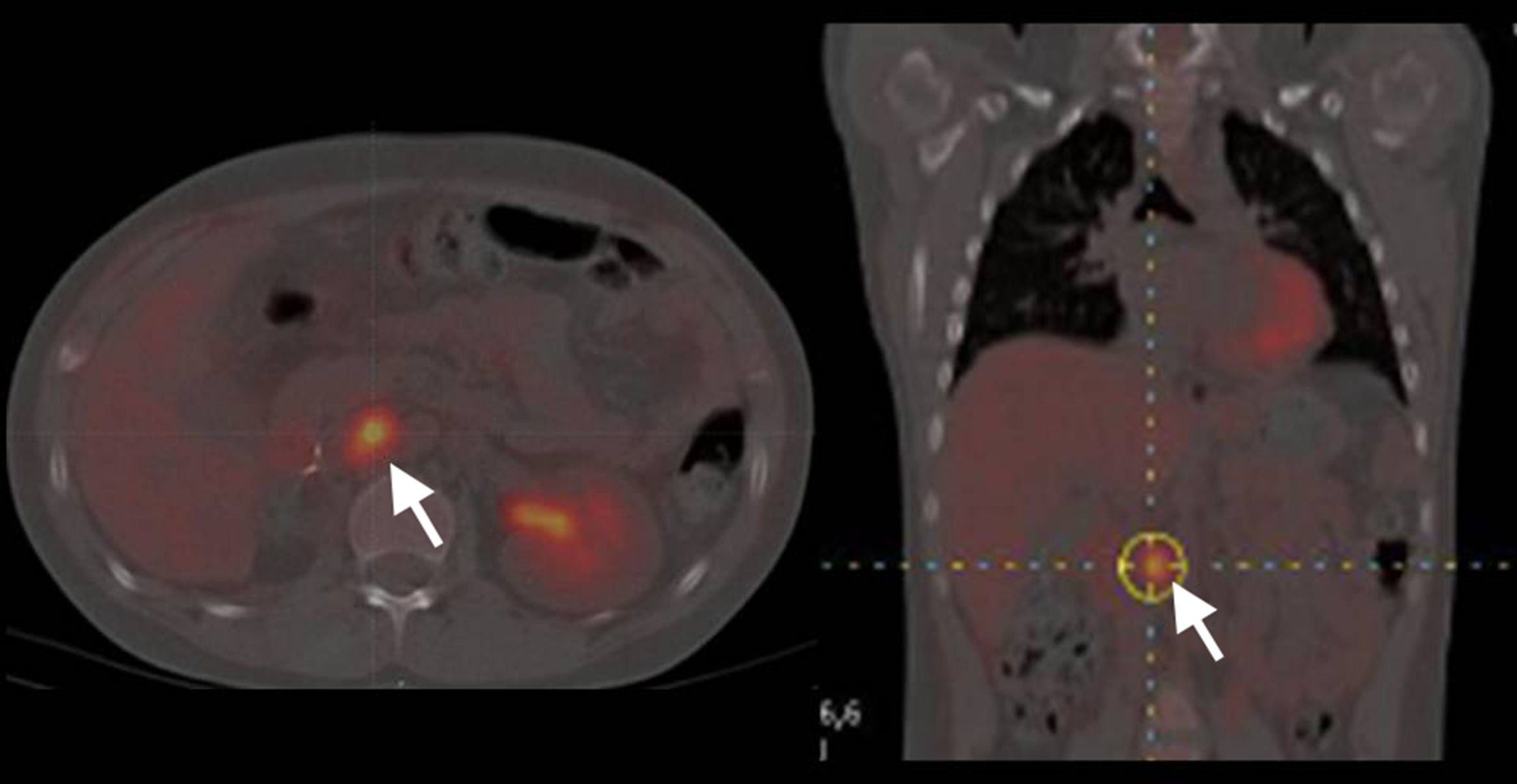
Comparación de técnicas diagnósticas en un caso de feocromocitoma. El estudio mediante SPECT empleando como trazador 123I-MBG no muestra captación patológica (A). En contraste, el estudio mediante PET empleando como trazador 18-fluorodesoxiglucosa en la misma paciente muestra una captación heterogénea correspondiente a un tumor de gran tamaño en la glándula suprarrenal derecha (B) (flecha). Reproducido con autorización del Dr. Pacak25.
Ante la sospecha de FC/PG con resultados bioquímicos positivos debe realizarse inicialmente una TC/RM, seguida de imágenes funcionales con [123I]-MIBG antes de la cirugía. Si se dispone de PET la utilización de 6-[18F]-fluordopamina o [18F]-DOPA puede substituir a [123I]-MIBG. En caso de que las escintigrafías con trazadores específicos fallen en la localización del tumor y/o se deba excluir la presencia de tumores malignos o metástasis antes de la cirugía, podría realizarse PET con [18F]-FDG o escintigrafía convencional con octreótido marcado.
MolecularNecesario en los pacientes en edad pediátrica, independientemente de su historia familiar, siendo los genes más relevantes, por la frecuencia de afectación en este periodo de la vida: VHL, en el caso de FC, y SDHB en el caso de PG o tumores malignos. La secuencia de genes a estudiar en FC/PG aparentemente esporádicos se basa, en el caso de los niños, en la localización del tumor y en el patrón de secreción de catecolaminas:
- -
FC noradrenérgico: VHL, SDHB, SDHD y SDHC.
- -
FC adrenérgico: RET y SDHx.
- -
PG abdominal/torácico simpático: VHL, SDHB, SDHD y SDHC.
- -
PG de la cabeza/cuello parasimpático: SDHD, SDHB, SDHC, VHL y SDH5.
Solo alrededor del 12% de los FC/PG en la edad pediátrica son malignos (incidencia de 0,02 casos/106 niños/año). No existen imágenes histológicas o inmunohistoquímicas capaces de predecir el potencial metastásico de este tipo de tumores. Por consiguiente, la biopsia preoperatoria no solo no está indicada, sino que es potencialmente peligrosa. La malignidad se establece por la presencia de metástasis a distancia en una localización donde los paraganglios no se localizan habitualmente (ganglios, hígado, pulmón y hueso, fundamentalmente). El riesgo de malignidad es mayor en los tumores de más de 5cm, en los PG simpáticos más que en los FC o en los PG de la cabeza-cuello y, especialmente, en los PG simpáticos por mutaciones en SDHB. Alrededor del 50% de los tumores malignos lo son por mutaciones en SDHB. También, las mutaciones en MAX parecen conllevar un alto riesgo de malignidad, aunque dado su reciente descubrimiento la experiencia es escasa. La supervivencia media a los 5 años es del 36-74%, tanto en niños como adultos.
PronósticoEl riesgo de malignidad es mayor en los tumores de más de 5cm, en los PG simpáticos más que en los FC o en los PG de la cabeza-cuello y, especialmente, en los PG simpáticos por mutaciones en SDHB. Alrededor del 50% de los tumores malignos lo son por mutaciones en SDHB. También las mutaciones en MAX parecen conllevar un alto riesgo de malignidad. Una publicación reciente refiere 16 mutaciones patogénicas de MAX detectadas en 23 sujetos índice. Todos ellos tenían tumores suprarrenales, incluyendo 13 bilaterales o múltiples en la misma glándula. El 37% tenían antecedentes familiares. Dos pacientes desarrollaron enfermedad metastática. Los tumores por mutaciones de MAX se caracterizaron por presentar un incremento sustancial de normetanefrina, asociada con niveles normales o ligeramente elevados de metanefrina34. Los autores estiman que las mutaciones en MAX son responsables del 1,12% de FC/PG en pacientes sin evidencia de otras mutaciones.
La supervivencia media a los 5 años oscila entre el 35-75%, tanto en niños como adultos, pero estos datos deben ser analizados con más rigor cuando se conozca mejor la etiología de los tumores.
TratamientoTratamiento médicoEl tratamiento es fundamentalmente quirúrgico. No obstante, la cirugía conlleva un alto riesgo cardiovascular debido:a) a la liberación masiva de catecolaminas por la inducción anestésica, la intubación, las maniobras quirúrgicas o la manipulación del tumor (crisis hipertensiva, hemorragia cerebral, arritmia o infarto miocárdico); yb) a la hipotensión y shock, secundarios a la hipovolemia previa y a la caída brusca de catecolaminas tras el aislamiento (pinzamiento vascular) y extirpación del tumor; se recomienda que todos los pacientes con FC/PG, incluso aquellos con niveles aparentemente normales de catecolaminas, reciban el adecuado tratamiento previo a la cirugía (1-3 semanas) que permita bloquear los efectos de la liberación de catecolaminas.
Un bloqueador alfa es el fármaco inicial de elección, y el más empleado es la fenoxibenzamina (bloqueador alfa irreversible no competitivo) a dosis iniciales de 10mg cada 12h, con incrementos de 10-20mg cada 2-3 días hasta que se consiga un control adecuado de la tensión arterial (la dosis total habitual es de 1mg/kg/día), a menos que se produzca hipotensión arterial. También puede emplearse prazosina, doxazosina o terazosina.
Suele ser necesario seguir con un bloqueador beta si los pacientes presentan síntomas clínicos de estimulación del receptor beta (palpitaciones, ansiedad y dolor torácico). A tal efecto suele emplearse propanolol o atenolol para controlar la taquicardia refleja. También se recomienda incrementar el aporte de cloruro sódico y agua para favorecer la expansión del volumen plasmático y prevenir la hipotensión postoperatoria. La presencia de un anestesista experto y una estricta monitorización directa de la presión arterial y de la presión venosa central es obligada durante la intervención. Después del pinzamiento vascular del tumor puede producirse una profunda hipotensión que suele responder a una expansión rápida de la volemia con los fluidos apropiados38.
Tratamiento quirúrgicoConsiste en la extirpación completa del tumor (suprarrenalectomía y/o paragangliectomía), lo que permite la curación completa en más del 90% de los casos. En la última década, en el caso de los FC, la cirugía suprarrenal ha incluido técnicas mínimamente invasivas: suprarrenalectomía laparoscópica (transabdominal o retroperitoneal), restringida habitualmente a tumores <6cm y la tendencia, en el caso de tumores bilaterales o familiares con alto riesgo de recidiva contralateral, a preservar la función suprarrenal cortical mediante suprarrenalectomía subtotal en al menos una de las 2 suprarrenales, o autotrasplante heterotópico de corteza suprarrenal, aunque ello suponga dificultades técnicas y un mayor riesgo de recurrencia (10-38%).
Tratamiento del feocromocitoma/paraganglioma malignoLa extirpación de las recurrencias o de las metástasis es la principal opción y la única curativa. En los casos no susceptibles de cirugía, o en los que se requiere tratamiento adicional, las posibles opciones paliativas serían una combinación de: cirugía reductora de la masa tumoral, tratamiento con 131I-MIBG (400-500 mCi o dosis mayores asociadas a trasplante de médula ósea), quimioterapia o embolización tumoral, entre otros. Más recientemente han comenzado a evaluarse nuevas terapias: isótopos emisores de radiación ß o quimioterapia acoplados a análogos de somatostatina o inhibidores orales de la tirosincinasa.
SeguimientoLos niños diagnosticados de FC/PG deben seguirse de por vida con valoraciones anuales de: tensión arterial, metanefrinas en plasma y orina, cromogranina A y pruebas de imagen, preferentemente RM. También es necesario vigilar periódicamente a los pacientes portadores de mutaciones que confieran susceptibilidad para el desarrollo de estos tumores38.
Consideraciones finales- •
Los tumores de la corteza y médula suprarrenal, así como los PG, son raros en la infancia.
- •
En todos ellos la predicción de benignidad o malignidad es difícil. En el caso de los corticosuprarrenalomas la clasificación por estadios (i-iv) es el elemento más empleado en la actualidad para valorar el pronóstico. Los FC son en esencia benignos, pero la existencia de un porcentaje de malignidad hace que todo paciente requiera seguimiento individualizado y descartar la existencia de metástasis.
- •
La clasificación y patogenia de los tumores corticosuprarrenales son complejas.
- •
La genética ha aportado mucha información en los últimos años, conociéndose en la actualidad la importancia de las mutaciones en el gen TP53 en los tumores de la corteza suprarrenal, del PRKAR1A en el complejo de Carney y la existencia de al menos 10 genes cuyas mutaciones pueden generar feocromocitomas/paragangliomas: VHL, RET, NF1, SDHA, SDHAF2, SDHB, SDHC, SDHD,TMEM127 y MAX, presentando, en general, un mayor grado de malignidad los pacientes con mutaciones en los genes SDHB y MAX.
- •
Las técnicas de imagen han mejorado mucho el proceder diagnóstico, incluyendo además de la TC y la RM las técnicas de imagen funcional como la gammagrafía corporal total con MIBG marcada con 123I o con 131I de alta sensibilidad y especificidad, la SPECT y la PET. La PET genera menor exposición a radiación y mayor rapidez en la obtención de imágenes, pudiendo utilizarse con [18F]- FDG o con trazadores más específicos para tejido cromafín ([18F]-DOPA o 6-[18F]-fluordopamina). La PET ([18F]-DOPA es más sensible que la MIBG-123I. Por consiguiente, se prefiere el empleo de la PET en el diagnóstico de los FC. Si no se dispone de ella, emplear MIBG-123I en caso de FC suprarrenales no metastáticos y gammagrafía para el receptor de somatostatina (SRS) en caso de FC con metástasis.
- •
La determinaciones de metanefrinas libres en sangre u orina son los marcadores bioquímicos de mayor importancia en el diagnóstico de FC/PG.
- •
El pronóstico es altamente variable y difícilmente predecible, por lo que cada paciente debe evaluarse de forma individualizada.
- •
El tratamiento en todos estos tumores es fundamentalmente quirúrgico, pudiendo desempeñar una función relevante la quimioterapia en los corticosuprarrenalomas (fundamentalmente en estadios iii y iv). En los FC/PG el tratamiento médico antihipertensivo peoperatorio e intraoperatorio es obligado (bloqueador alfa [fenoxibenzamina], seguido de un bloqueador beta [propanolol]).
- •
Todos los pacientes deberán seguir controles ulteriores y se les practicarán estudios analíticos y de imagen.
Los autores declaran no tener ningún conflicto de intereses.
