


Sr. Editor:
El agrandamiento hipofisario secundario a hipotiroidismo primario es bien conocido pero poco referido en la bibliografía médica, y menos aún en el ámbito pediátrico. La mejora sustancial de las técnicas de imagen desarrolladas en las dos últimas décadas (resonancia magnética [RM] y tomografía computarizada [TC] de alta resolución) revela que debe tratarse de una entidad más frecuente de lo que está descrito, si bien todavía son frecuentes las intervenciones quirúrgicas que tienen por objeto eliminar una masa hipofisaria que no se corresponda con un prolactinoma (tratable médicamente), y que, con gran probabilidad, dejan secuelas endocrinológicas permanentes.
El presente caso constituye una aportación más a la casuística de lo que se denomina hiperplasia tirotropa, una causa de tumor hipofisario tratable médicamente.
Niña de 8 años y 11 meses que es remitida desde otro centro hospitalario para intervención neuroquirúrgica de tumoración hipofisaria que se diagnostica con motivo de un cuadro de cefalea frontal de 1–2 meses de evolución y características orgánicas (nocturna y progresiva aunque en ausencia de vómitos). En la anamnesis dirigida refieren encontrarla, desde hace 4 meses, más irritable, asténica e inapetente, con un hábito intestinal algo más estreñido de lo habitual. No se encuentra poliuria ni polidipsia. Los antecedentes personales y familiares no revelan nada reseñable. En la exploración física destacan peso y talla en −0,72 desviación estándar (DE) (percentil 24) con un índice de masa corporal en −0,57 DE (percentil 29), y una palidez y sequedad cutáneas llamativas. No hay bocio visible ni aparentemente palpable. La auscultación cardiopulmonar y del abdomen no presenta alteraciones. Desarrollo puberal infantil (Tanner I). Neurológico rigurosamente normal.
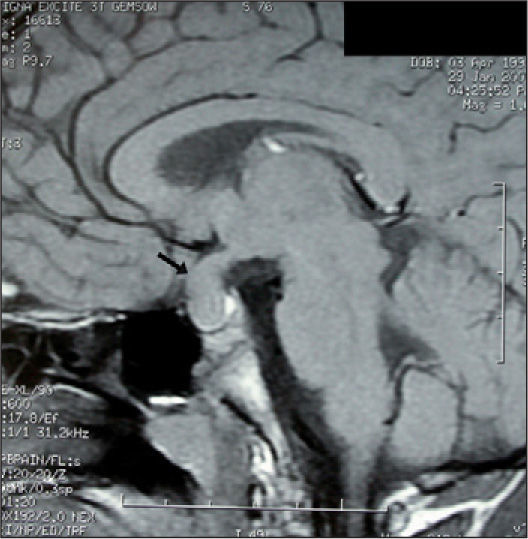
La RM que aportan es informada como “tumoración hipofisaria de 16mm × 18mm con extensión supraselar, sugestiva de macroadenoma” (fig. 1). También se aporta estudio analítico y hormonal, con alteraciones que confirman nuestro laboratorio: tirotropina (TSH) 1.117 μU/ml (valor normal [VN]: 0,25-6,15), tiroxina libre (T4L) 0,1ng/dl (VN: 0,7-1,64), T3 0,41ng/ml (VN: 0,6-1,8), anticuerpos antimicrosomales 156 U/ml y antitiroglobulina > 4.000 U/ml (VN: < 60), prolactina 67,59ng/ml (VN: < 20),
CPK 355 UI/l (VN: < 150) y colesterol total 442mg/dl. Se evalúa el resto de los ejes sin que se encuentren alteraciones, salvo IGF-1 65ng/ml (percentil 5).
Con el diagnóstico de hipotiroidismo primario, se realiza una ecografía tiroidea y una ecocardiografía, que objetivan un aumento generalizado del tiroides con incremento llamativo de la vascularización y derrame pericárdico leve sin repercusión hemodinámica. La campimetría visual no revela alteraciones y la edad ósea se encuentra retrasada (7 años).
Se establece el diagnóstico de hipotiroidismo primario grave por tiroiditis autoinmune con hiperplasia tirotropa, hiperprolactinemia, derrame pericárdico, hipercolesterolemia y elevación de la CPK, muy probablemente secundarios. Con ello se desestima la indicación quirúrgica y se inicia tratamiento con levotiroxina en dosis sustitutivas (4μg/kg/día); pasado un mes, se repite el control analítico, ecocardiográfico y de RM: TSH 1,48 μU/ml, T4L 2,11ng/dl, prolactina 11,2ng/ml, colesterol total de 154mg/dl, CPK 87 UI/l; el derrame pericárdico ha desaparecido pero persiste la tumoración hipofisaria. La sintomatología referida al inicio desaparece por completo.
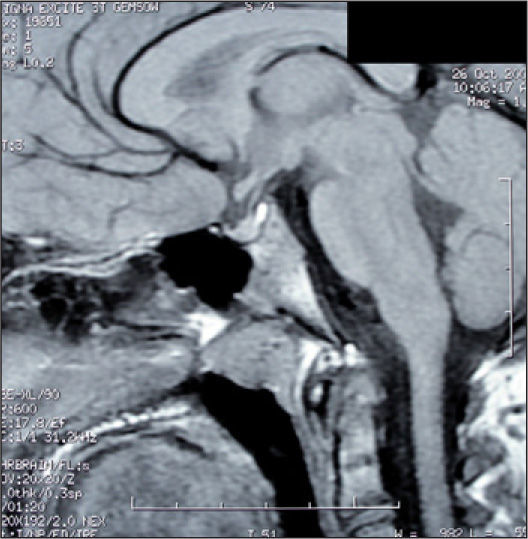
En la evolución, controles analíticos sucesivos siguen mostrando normalidad de los parámetros anteriormente mencionados, y en una tercera RM realizada 3 meses después se revela una reducción significativa del tamaño tumoral, que desaparece por casi por completo con la realizada a los 9 meses del diagnóstico (fig. 2).
El agrandamiento hipofisario compensatorio se ha descrito en situaciones de insuficiencia suprarrenal primaria, hipogonadismo primario e hipotiroidismo primario1. Esta última entidad, actualmente denominada más correctamente como hiperplasia tirotropa, se describió por primera vez en el año 1851 en pacientes con cretinismo2. Desde entonces, los casos publicados han sido relativamente numerosos en adultos pero escasamente referidos en la edad pediátrica1–3. La causa de hiperplasia tirotropa más frecuentemente detectada, tanto en niños como en adultos, ha sido la de tiroiditis crónica autoinmune1,4,5, hallazgo que coincide con nuestra aportación; la etiología congénita (agenesia/hipoplasia tiroidea o dishormonogénesis), aunque globalmente más frecuente, ha sido menos referida en las últimas décadas con motivo de la detección precoz de hipotiroidismo congénito6
La clínica predominante de estos pacientes es la de hipotiroidismo, habitualmente grave, aunque en algún caso, como el nuestro, no excesivamente sintomático3. Los síntomas dependientes de la compresión local son los comúnmente descritos a los de cualquier otro origen, esto es, deterioros del campo visual, hipertensión intracraneal, los derivados de déficits hormonales y la hiperprolactinemia secundaria1,3. El tamaño de la porción tirotropa que justifica el efecto compresivo mencionado confiere a la hipófisis dimensiones variables, desde pocos milímetros hasta casos que simulan macroadenomas y, como en nuestro caso, tienen extensión supraselar1. La presencia de retraso puberal y del crecimiento descritos en los casos pediátricos probablemente resultan de la conjunción de todos estos factores (hipotiroidismo grave, déficits hormonales y/o hiperprolactinemia)3.
En el diagnóstico diferencial destaca el adenoma hipofisario productor de TSH, un 1-2 % de todos los adenomas pituitarios, en el que, además, coexista la situación de hipotiroidismo primario5. Aunque infrecuentes, tales casos parecen consecuencia de un proceso que acaba haciendo irreversible una situación prolongada de hiperplasia tirotropa. En este estado se puede comprobar como el tratamiento sustitutivo con levotiroxina regulariza o incrementa las concentraciones de T4 libre con imposibilidad de normalizar las de TSH5,7. Por otra parte, aunque la hiperprolactinemia constituye un hallazgo frecuente en la hiperplasia tirotropa por exceso compensatorio de hormona liberadora de tirotropina (TRH) y/o compresión de tallo, existen casos de coexistencia con prolactinoma en los que tampoco la elevación de la prolactina se normaliza tras regularizarse la función tiroidea5. Por último, no debe olvidarse la excepcional resistencia generalizada a hormonas tiroideas en la que, si bien existe una elevación de hormonas tiroideas, la clínica es de hipotiroidismo franco con incremento compensatorio de TSH y, en algún caso, hiperplasia tirotropa8.
El tiempo de respuesta terapéutica necesario para la regresión de la masa hipofisaria es ampliamente variable, desde casos en los que se refiere una remisión tumoral en apenas 6 días9 hasta otros, como el nuestro y la mayoría, en los que ésta se constata varias semanas o meses después.
Aunque la cirugía de esta entidad no está indicada, existen unos pocos casos publicados en los que se realiza como primer procedimiento10. En este sentido, y como pretexto para algunos autores de este inadecuado proceder, resulta útil el empleo intraoperatorio de técnicas de análisis clonal sobre la pieza quirúrgica que permita la diferenciación entre hiperplasia tirotropa y adenoma hipofisario4,5. Otros autores teorizan con criterios que permitirían de un modo incruento una fácil diferenciación entre ambas entidades, a saber, la relación α-TSH/TSH y el grado de actividad para el radiotrazador 18-fluorodesoxiglucosa (18FDG) en la tomografía por emisión de positrones (PET)-TC7. No obstante, sólo la deducción clínica y, de forma excepcional, el uso de este tipo de técnicas, podrán discernirlas para evitar secuelas de una cirugía transesfenoidal4; tampoco ayudan las técnicas de imagen de alta resolución como la RM y la TC10.
A modo de conclusión, este caso recuerda la importancia de un estudio basal de todos los ejes hormonales en toda tumoración de origen selar que excluya las causas no quirúrgicas, a saber, el prolactinoma, el hipogonadismo hipergonadotropo, la insuficiencia suprarrenal primaria y la aparentemente menos conocida hiperplasia tirotropa secundaria a hipotiroidismo primario.
