









El trasplante de progenitores hematopoyéticos (TPH) consiste en implantar elementos celulares capaces de generar un sistema hematopoyético nuevo y sano. El régimen de intensidad reducida (RIR) consiste en un tratamiento predominantemente inmunosupresor, para facilitar un implante progresivo con menor morbilidad. Este tipo de acondicionamiento puede también provocar mielosupresión, aunque potencialmente reversible en el tiempo.
El acondicionamiento RIR permite aplicar TPH a pacientes con enfermedad genética en los que no es deseable añadir comorbilidad por las altas dosis de quimioterapia que conlleva el régimen mieloablativo convencional.
Pacientes y métodosSe analiza la evolución de 68 pacientes pediátricos con enfermedades genéticas que entre los años 2005-2013 se han sometido a un TPH con RIR en las Unidades pediátricas de Trasplante Hematopoyético de los hospitales españoles integrantes del Grupo Español para Trasplante de Médula Ósea en niños.
Se trata de un estudio multicéntrico que incluye a 68 pacientes, de los cuales 43 presentan inmunodeficiencia primaria, 21 presentan hemopatía congénita y 4 están afectados de metabolopatía.
ResultadosCincuenta de los 68 pacientes se encuentran vivos (73,5%). La supervivencia global (SG) a 9 años es de 0,74. Veintitrés (33,8%) han presentado en el transcurso del TPH algún evento. Supervivencia libre de evento de 0,66. La SG en los pacientes con hemopatía es de 0,81; en las inmunodeficiencias primarias es de 0,70 y en las metabolopatías es de 0,4. No se observa diferencia significativa entre los 3 grupos de enfermedades. Respecto a la fuente de progenitores hematopoyéticos, la SG en los pacientes trasplantados con sangre periférica es de 0,74; con médula ósea es de 0,70 y con la sangre de cordón umbilical es de 0,70. No se observa tampoco diferencia estadística significativa.
ConclusionesEn nuestro trabajo, de ámbito nacional, hemos evidenciado unos resultados favorables en TPH con régimen de intensidad reducida en las enfermedades genéticas. Cabe destacar que las metabolopatías requieren una consideración individualizada para sopesar en cada paciente los riesgos y beneficios que comporta el RIR.
Haematopoietic stem cell transplantation (HSCT) involves implanting cellular elements capable of generating a new and healthy haematopoietic system. Reduced intensity conditioning (RIC) consists of an immunosuppressive treatment to facilitate a progressive implant with lower morbidity. This type of conditioning can also lead to myelosuppression, which is potentially reversible over time.
Reduced intensity conditioning enables HSCT to be performed on patients with genetic diseases for whom added comorbidity is undesirable due to the high doses of chemotherapy that accompanies conventional myeloablative regimens.
Patients and methodsAn analysis was performed on the outcomes of 68 paediatric patients with genetic diseases who underwent HSCT with RIC between 2005 and 2013 in the of Paediatric Haematopoietic Stem Cell Transplantation Units that are part of the Spanish Working Group for Bone Marrow Transplantation in Children.
A multicentre study was conducted including 68 patients, of whom 43 had Primary Immunodeficiency, 21 with congenital haematological diseases, and 4 with metabolic diseases.
ResultsFifty (73.5%) of the 68 patients were still alive. The Overall Survival (OS) at nine years was 0.74. Twenty-three (33.8%) had some event during the course of the HSCT, with an event-free survival rate of 0.66. The OS in patients with haematological diseases was 0.81, being 0.7 in primary immunodeficiencies, and 0.4 in metabolic diseases. No significant difference was observed between the 3 groups of diseases. As regards the source of haematopoietic progenitors, there was an OS rate of 0.74 in patients transplanted with peripheral blood, 0.70 with bone marrow, and 0.70 and with cord blood, with no statistically significant differences.
ConclusionsFavourable results have been obtained in HSCT with reduced intensity conditioning in genetic diseases. It should be noted that the risks and benefits of the RIC in patients with metabolic diseases need to be assessed on an individual basis.
El trasplante de progenitores hematopoyéticos (TPH) consiste en implantar elementos celulares capaces de generar un sistema hematopoyético nuevo y sano. Los progenitores hematopoyéticos (PH) son células multipotenciales, capaces de diferenciarse, multiplicarse y autoperpetuarse en distintas líneas celulares. El término régimen de acondicionamiento se refiere al tratamiento que se administra al paciente antes de la infusión de los PH.
El régimen de intensidad reducida (RIR) consiste en un tratamiento de acondicionamiento predominantemente inmunosupresor para facilitar un implante progresivo con menor morbilidad. Este tipo de acondicionamiento puede también provocar mielosupresión, aunque potencialmente reversible en el tiempo. Una vez implantadas las células del donante, suele ocurrir quimerismo mixto (coexistencia de células derivadas del donante y del receptor en el mismo espacio medular) que puede estabilizarse o evolucionar a quimera total o rechazo.
El régimen de acondicionamiento de intensidad reducida permite aplicar el TPH a pacientes con enfermedad genética en los que no es deseable añadir comorbilidad por las altas dosis de quimioterapia que conlleva el régimen mieloablativo convencional.
Según el Center of International Blood and Marrow Research1, los regímenes de acondicionamiento de intensidad reducida están basados en el uso de dosis altas de fármacos inmunosupresores, especialmente fludarabina (Flu) y globulinas antilinfocitarias, combinados con dosis menores de fármacos alquilantes (busulfán, melfalán, treosulfán [Tre], ciclofosfamida o tiotepa [TT]). La irradiación corporal total (ICT) tiene como propósito causar ablación hematopoyética e inmunosupresión.
Los acondicionamientos RIR son los que incluyen:
- –
ICT ≤ 500 cGy en dosis única o ≤ 800 cGy si es fraccionada.
- –
Busulfán: dosis total ≤ 9mg/kg.
- –
Melfalán: dosis total ≤ 140mg/m2.
- –
TT: dosis total ≤ 10mg/kg.
- –
Ciclofosfamida: dosis total ≤ 120mg/kg.
- –
Tre: dosis total ≤ 42g/m2.
- –
Flu: dosis total ≤ 160mg/m2.
Se define implante o recuperación hematológica cuando el receptor recupera cifras de neutrófilos en sangre periférica (SP) ≥ 0,5×109/l y de plaquetas ≥ 20×109/l.
La reconstitución inmunitaria tras el TPH alogénico es un proceso lento, no alcanzándose valores de células T circulantes normales hasta los 6-12 meses después del trasplante; en consecuencia, la morbilidad durante el primer año post-TPH puede ser alta2.
Las enfermedades por las que reciben los niños un TPH son mayoritariamente neoplásicas, si bien este trabajo se limita a la aplicación en enfermedades genéticas, que son en las que tiene un mayor interés un RIR.
La β-talasemia mayor es la forma más grave de talasemia. Los pacientes presentan anemia diseritropoyética con dependencia transfusional, esplenomegalia, deformidades óseas y hemosiderosis. El TPH es actualmente el único tratamiento curativo. La experiencia con TPH de donante no emparentado (DnE) y con acondicionamiento de intensidad reducida todavía es escasa, siendo el problema principal de este tipo de acondicionamiento el rechazo del injerto en pacientes multitransfundidos3,4.
La drepanocitosis es otra hemoglobinopatía que cursa con crisis vasooclusivas en la que también el TPH alogénico es el único tratamiento curativo y se reserva para aquellos pacientes que presentan manifestaciones clínicas severas5.
La anemia de Fanconi es una enfermedad hereditaria que se caracteriza por pancitopienia, anomalías físicas y tendencia a neoplasia. Presentan fragilidad cromosómica con mayor sensibilidad a quimioterapia y radioterapia. Pese al tratamiento de soporte, la enfermedad es inevitablemente fatal una vez desarrollada la afectación medular. El trasplante alogénico de PH es el único tratamiento curativo disponible en la actualidad6,7.
Las inmunodeficiencias primarias (IDP) son un grupo heterogéneo de defectos congénitos que afectan a distintos mecanismos esenciales de la respuesta inmunológica. Se caracterizan por mayor propensión a infecciones, enfermedades autoinmunes y neoplasias. El TPH es el único tratamiento curativo para un amplio espectro de IDP8,9.
Las metabolopatías son enfermedades cuyo tratamiento es multidisciplinar y el TPH es una posible opción terapéutica en algunas de ellas. Los principales problemas del TPH en estas enfermedades son el fallo del implante con recuperación autóloga y la falta de donantes familiares histocompatibles10. La sangre de cordón umbilical (SCU) ha demostrado resultados favorables11.
Se presentan los resultados del TPH en enfermedades genéticas con régimen de acondicionamiento de intensidad reducida reportados en el Grupo Español para el Trasplante de Médula Ósea en Niños (GETMON).
Pacientes y métodosSe ha realizado un estudio multicéntrico, observacional, retrospectivo-prospectivo, descriptivo y analítico.
Los individuos sujetos al estudio son 68 pacientes pediátricos (entre 0 y 18 años) con enfermedades genéticas sometidos a TPH con régimen de acondicionamiento de intensidad reducida, entre los años 2005-2013, en las Unidades Pediátricas de Trasplante Hematopoyético de los hospitales españoles integrantes del GETMON. Los investigadores de los centros participantes completaron una plantilla de recogida de datos. Los tutores legales de los pacientes han firmado un consentimiento informado del TPH y de los estudios subsidiarios.
En el estudio estadístico, las curvas de supervivencia global (SG) y supervivencia libre de evento (SLE) fueron realizadas siguiendo el método de Kaplan-Meier y comparadas por el test log-rank. El nivel de significación estadístico se consideró cuando la p ≤ 0,05. El análisis de los datos se ha realizado con el programa estadístico SPSS (Statistical Package for Social Sciences) versión 22.0 para Windows.
Se consideran evento los acontecimientos desfavorables que ocurren durante el período posterior al TPH, tales como fallecimiento y fallo del implante. Se define como SG el tiempo transcurrido entre la realización del TPH y el último seguimiento. Se define como SLE el tiempo transcurrido entre la realización del TPH y la presentación de cualquier evento.
ResultadosDescripción de los pacientesDel total de 68 pacientes, 40 (59%) eran niños y 28 (41%) niñas. La mediana de edad al diagnóstico fue de 21 meses (intervalo: 10 días-180 meses). La mediana de edad al trasplante fue de 46 meses (intervalo: 3 meses-190 meses).
Cuarenta y tres pacientes estaban afectados de IDP: inmunodeficiencia combinada grave (22), linfohistiocitosis hemofagocítica familiar (7), síndrome de hiper IgM ligado al cromosoma X (3), síndrome de Omenn (2), síndrome de Wiskott-Aldrich (2), deficiencia antígenos leucocitarios humanos (HLA)-II (2), síndrome IPEX (2), síndrome de Griscelli (1), déficit de adhesión de los leucocitos tipo 1 (1), y una IDP grave sin filiar. Veintiún pacientes presentaban hemopatía congénita: anemia de Fanconi (12), talasemia mayor (7), drepanocitosis (1) y un déficit de piruvato cinasa. Cuatro presentaban metabolopatías: mucopolisacaridosis (2), fucosidosis (1) y leucodistrofia globoide o enfermedad de Krabbe (1). Las IDP suponían un 63,2% del total de pacientes, las hemopatías congénitas un 30,9% y las metabolopatías un 7% (fig. 1).
Características del trasplante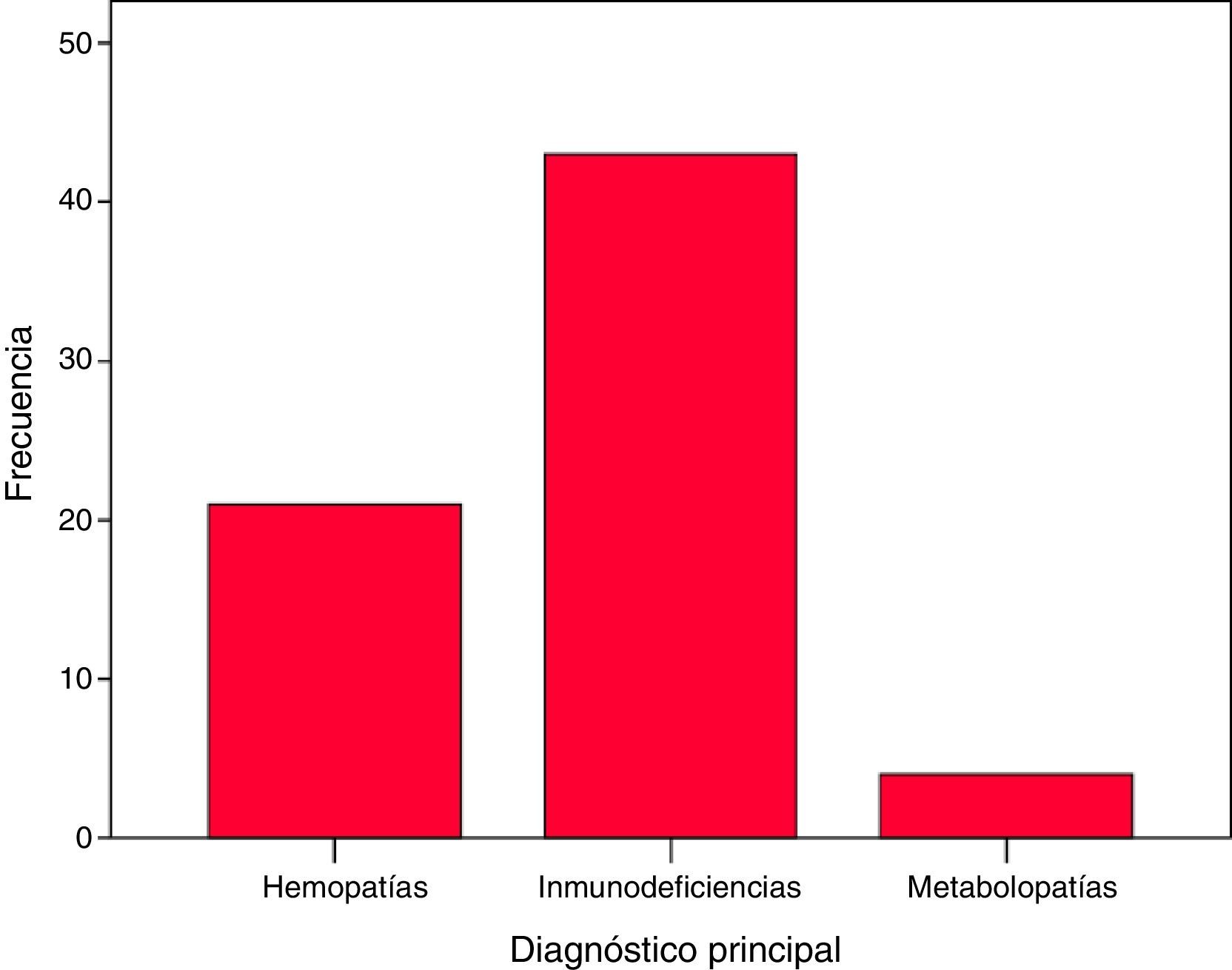
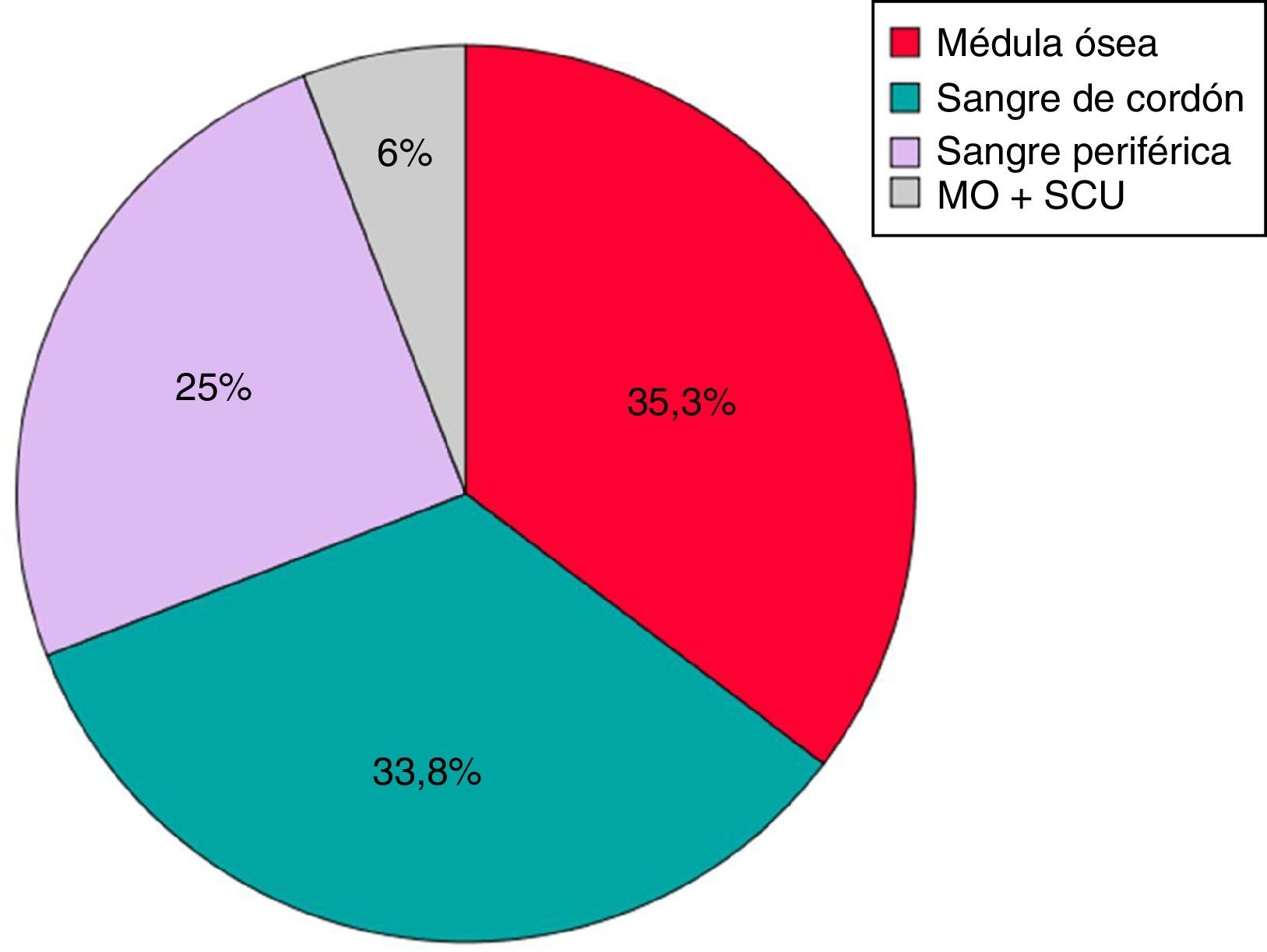
Según la fuente de PH, 24 trasplantes se realizaron a partir de médula ósea (MO) (35,3%), 23 de SCU (33,8%), 17 de SP (25%) y 4 combinados de SCU+MO (6%) (fig. 2).
Respecto al tipo de donante, 47 trasplantes (69,2%) fueron de DnE, 25 presentaban disparidad del tipificación HLA y en 22 el HLA era idéntico (presentaban identidad alélica en A B C DR (8/8)±DQ (10/10)). Veintiuno fueron a partir de donante familiar o emparentado (31%); entre estos últimos, 13 fueron de hermano HLA idéntico y 8 fueron de familiar HLA no idéntico (parental). Según la identidad HLA, encontramos 35 TPH con HLA idéntico (51,5%) y 33 con HLA no idéntico (48,5%).
El tratamiento de acondicionamiento pretrasplante se basó mayoritariamente en el uso de Flu, en concreto en 65 de los 68 pacientes. Los regímenes fueron Flu+melfalán (33); Flu+ciclofosfamida±ICT (13); Flu+Tre+TT (7); Flu±Tre±ciclofosfamida (6); Flu±busulfán±TT (7); Flu+melfalán+ICT (1) y busulfán+ciclofosfamida (1). En todos los casos, las dosis máximas estaban dentro de los límites aceptados como RIR.
En 57 pacientes se asociaron anticuerpos monoclonales. En 40 (58,8%) se administró gammaglobulina antitimocítica (GAT) con dosis total entre 6-10mg/kg y en 17 casos (25%) se administró alemtuzumab (anticuerpo anti-CD52) con dosis total de 1mg/kg.
La depleción de los linfocitos T se ha realizado en 14 casos: 6 de TPH de SP haploidéntico parental, 6 de SP de DnE HLA idéntico, uno de SP de hermano HLA idéntico y uno de MO de DnE con HLA idéntico.
Para la profilaxis de la enfermedad injerto contra huésped (EICH), en 29 pacientes se ha utilizado ciclosporina sola, en 26 pacientes la asociación de ciclosporina+micofenolato, y en 10 casos ciclosporina+metotrexato±micofenolato. Solo en 3 pacientes no se ha utilizado profilaxis y estos correspondían a TPH haploidénticos de madre con fuente de SP, en los que se realizó depleción de linfocitos T como único tratamiento profiláctico de EICH previamente a la infusión del trasplante.
Respecto a la celularidad de la infusión del TPH, de los 24 pacientes que recibieron MO, la media ± DE de células nucleadas (CN) infundidas fue de 19,12×108/kg±36,8×108/kg y la de células CD34+ infundidas fue de 11,51×106/kg±14,68×106/kg. De los 23 pacientes que recibieron SCU, la media ± DE de CN infundidas fue de 3,51×108/kg±5,1×108/kg y la de células CD34+ infundidas fue de 1,3×106/kg±2,12×106/kg. De los 17 pacientes que recibieron SP, la media ± DE de células CD34+ infundidas fue de 15,86×106/kg±17,83×106/kg. Cuatro pacientes recibieron combinación de SCU+MO de hermano HLA idéntico, la media ± DE de CN infundidas fue de 12,82×108/kg±21,2×108/kg y la de células CD34+ infundidas fue de 4,43×106/kg±2,04×106/kg.
Evolución postrasplanteSesenta y cinco pacientes (95,6%) han presentado implante de neutrófilos. Dichos pacientes han alcanzado una cifra de neutrófilos ≥ 0,5×109/l en una mediana de 17,4 días. Sesenta y un pacientes (89,7%) han presentado implante de plaquetas, alcanzado una cifra de plaquetas ≥ 20×109/l en una mediana de 24 días postrasplante.
El estudio de quimera ha mostrado quimera total (100% del donante) en linfocitos T y granulocitos entre los 30 y 60 días post-TPH en 38 pacientes (55,9%). En 26 pacientes (38,2%) se ha detectado quimera mixta y en 4 pacientes (5,9%) con fallo de implante primario se constató la recuperación autóloga.
De los 60 pacientes evaluables a ≥ 6 meses post-TPH para el estudio de quimera, se observó quimera total del donante para linfocitos T y granulocitos en 32 pacientes (47,1%). En 21 pacientes (30,9%) se ha constatado quimera mixta y estable en el tiempo, 2 pacientes (2,9%) presentaron fallo del implante primario y 5 pacientes (10,2%), que inicialmente presentaron quimera mixta, evolucionaron con fallo del implante secundario.
Respecto al estudio de la EICH, 24 pacientes no han presentado ningún grado de EICH aguda (35,3%), 33 han presentado EICH aguda grado i-ii (48,5%), 11 han presentado EICH aguda grado iii-iv (16,2%).
De los 64 pacientes evaluables para EICH crónica a 100 días post-TPH, 55 no han presentado EICH crónica (80,9%), 4 han presentado EICH crónica limitada y 5 han presentado EICH crónica extensa.
De los 18 pacientes fallecidos, las causas principales, aisladas o combinadas, fueron: EICH, sepsis, fallo multiorgánico, fallo del implante primario o secundario, hemorragia pulmonar, hemorragia cerebral o infección por citomegalovirus.
Estudio de supervivenciaEn el momento actual, 50 de los 68 pacientes se encuentran vivos (73,5%) (fig. 3).
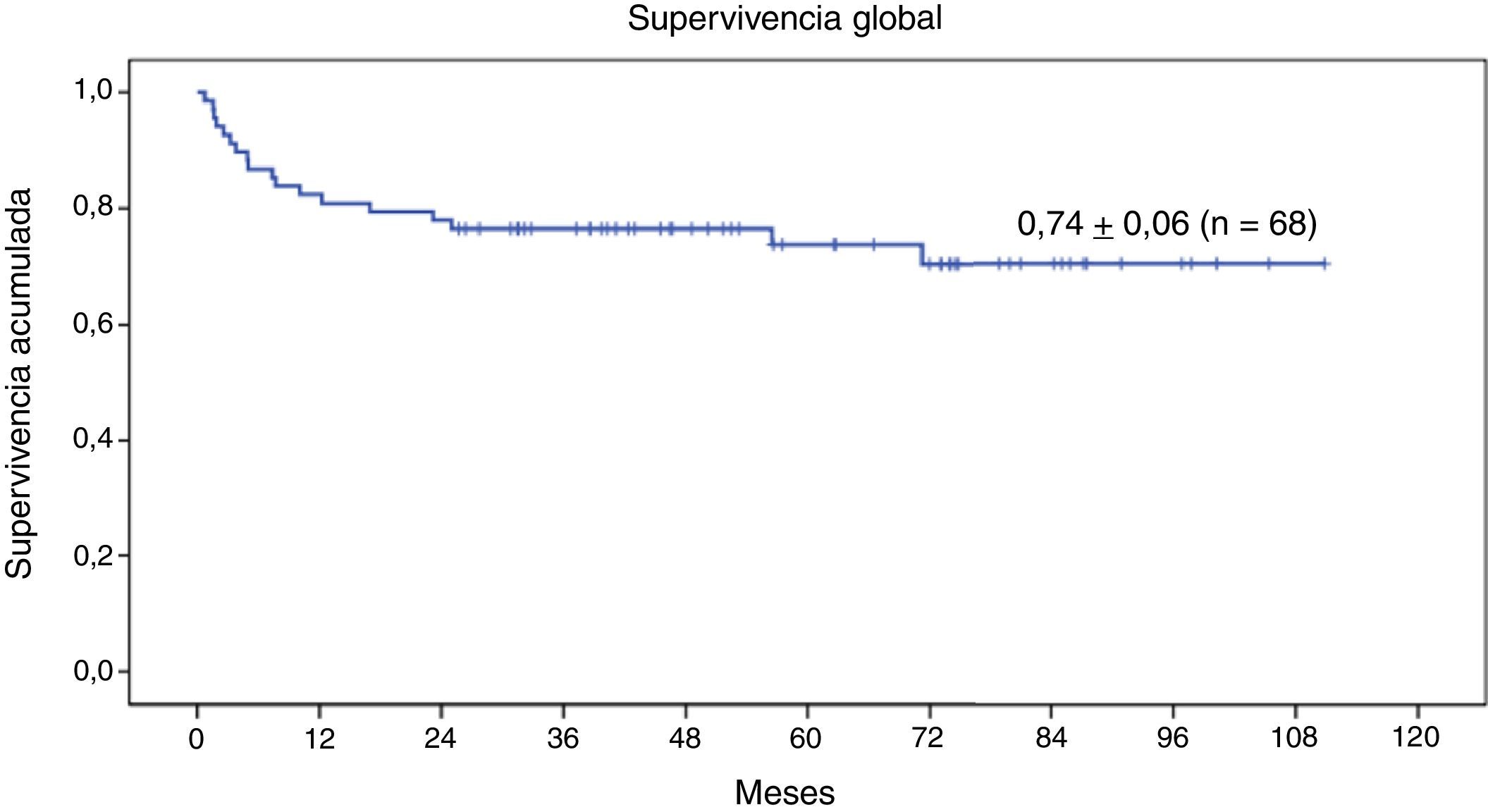
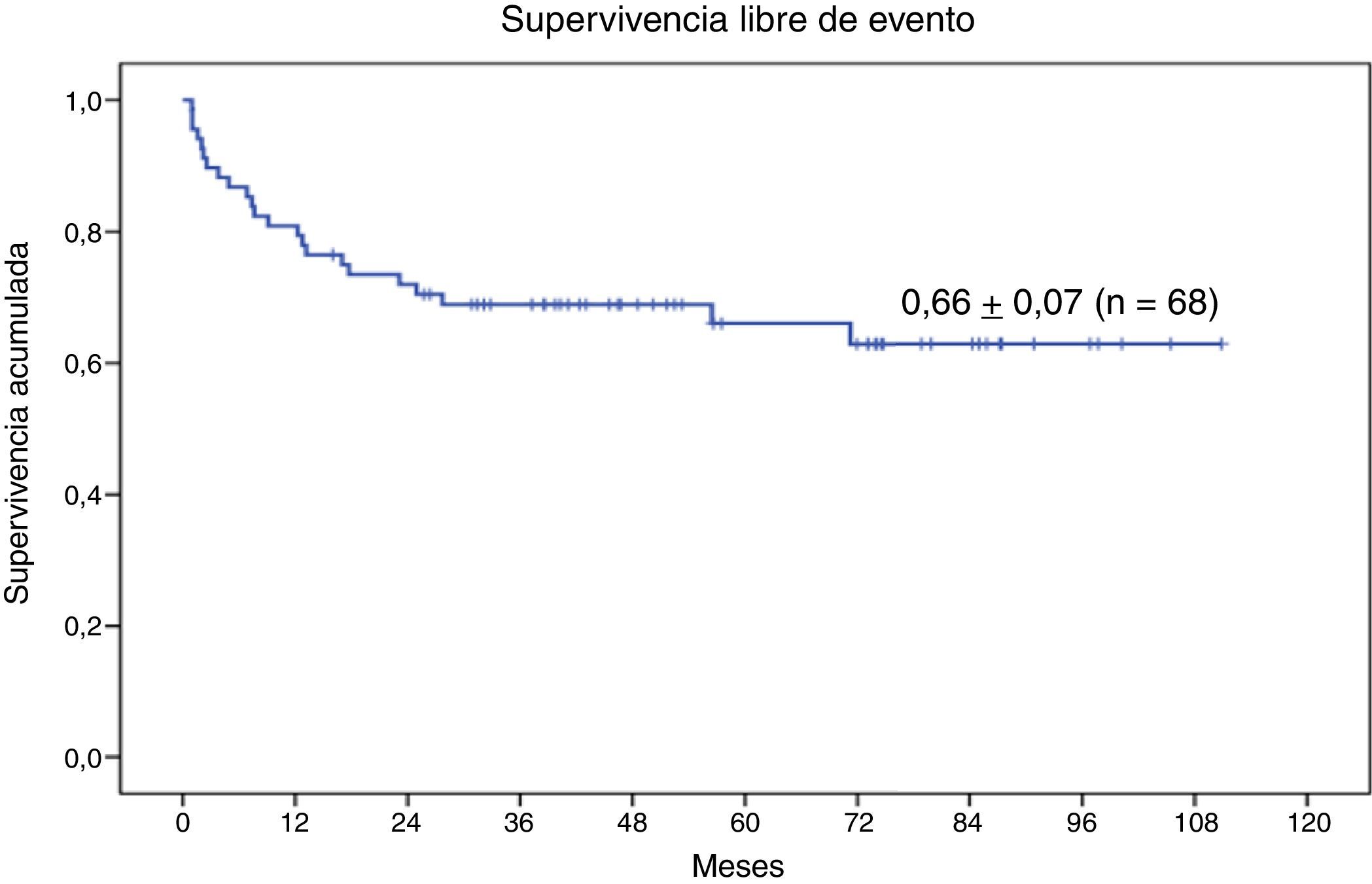
La SG a 9 años de la serie completa de 68 pacientes con enfermedad genética sometidos a TPH con intensidad reducida es de 0,74±0,06 (fig. 3). Veintitrés (33,8%) han presentado en el transcurso del TPH algún evento. La SLE a 9 años es de 0,66±0,07 (fig. 4).
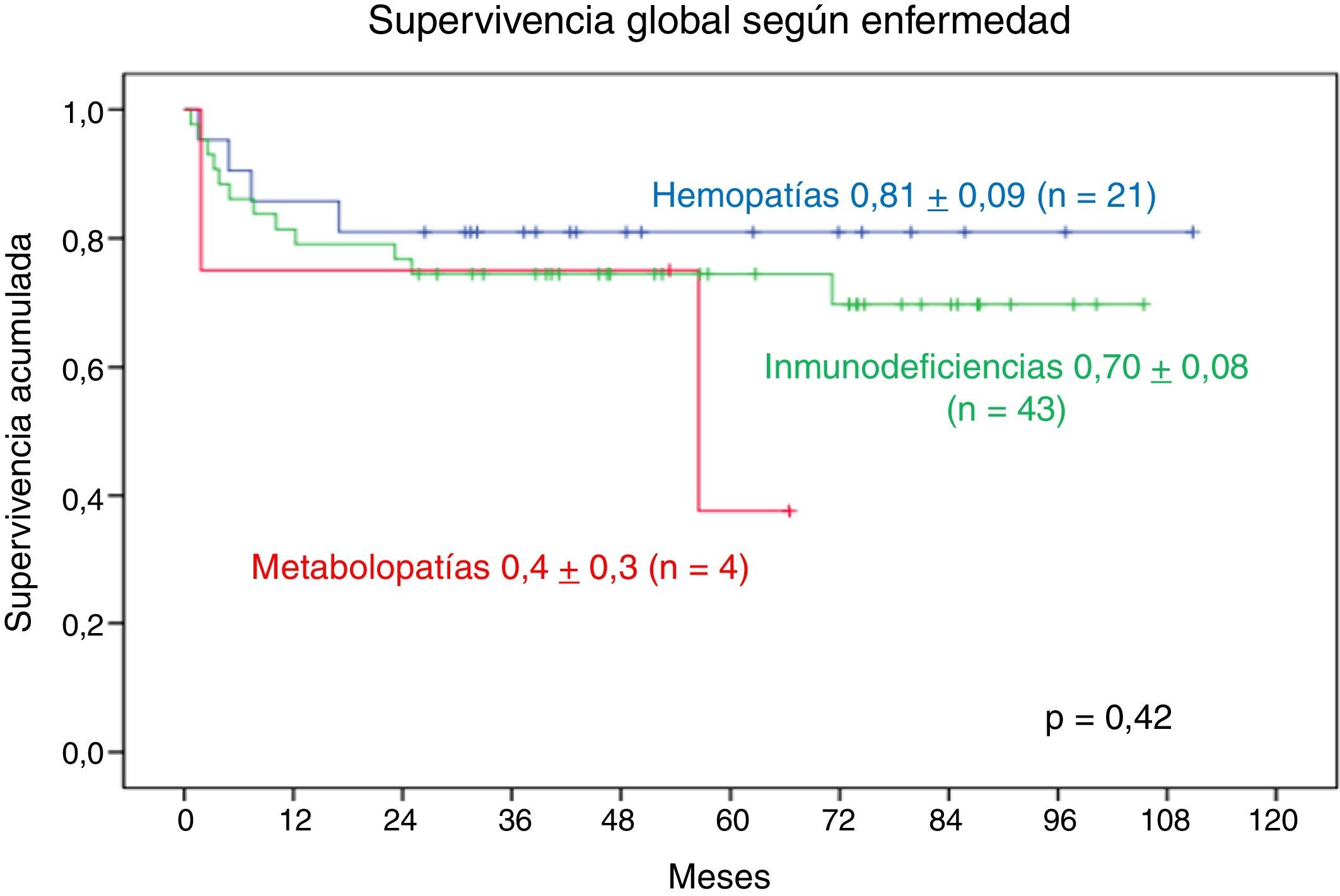
Comparando los 3 grupos principales de enfermedades, de los 21 pacientes con hemopatía, 17 continúan con vida; de los 43 pacientes con IDP, 31 continúan con vida, en tanto que de los 4 pacientes con metabolopatía, solo 2 continúan con vida.
La SG a 9 años en los pacientes con hemopatía es de 0,81±0,09, en los pacientes con IDP es de 0,70±0,08. La SG a 5 años en los pacientes con metabolopatía es de 0,4±0,3. No se observa diferencia significativa (fig. 5).
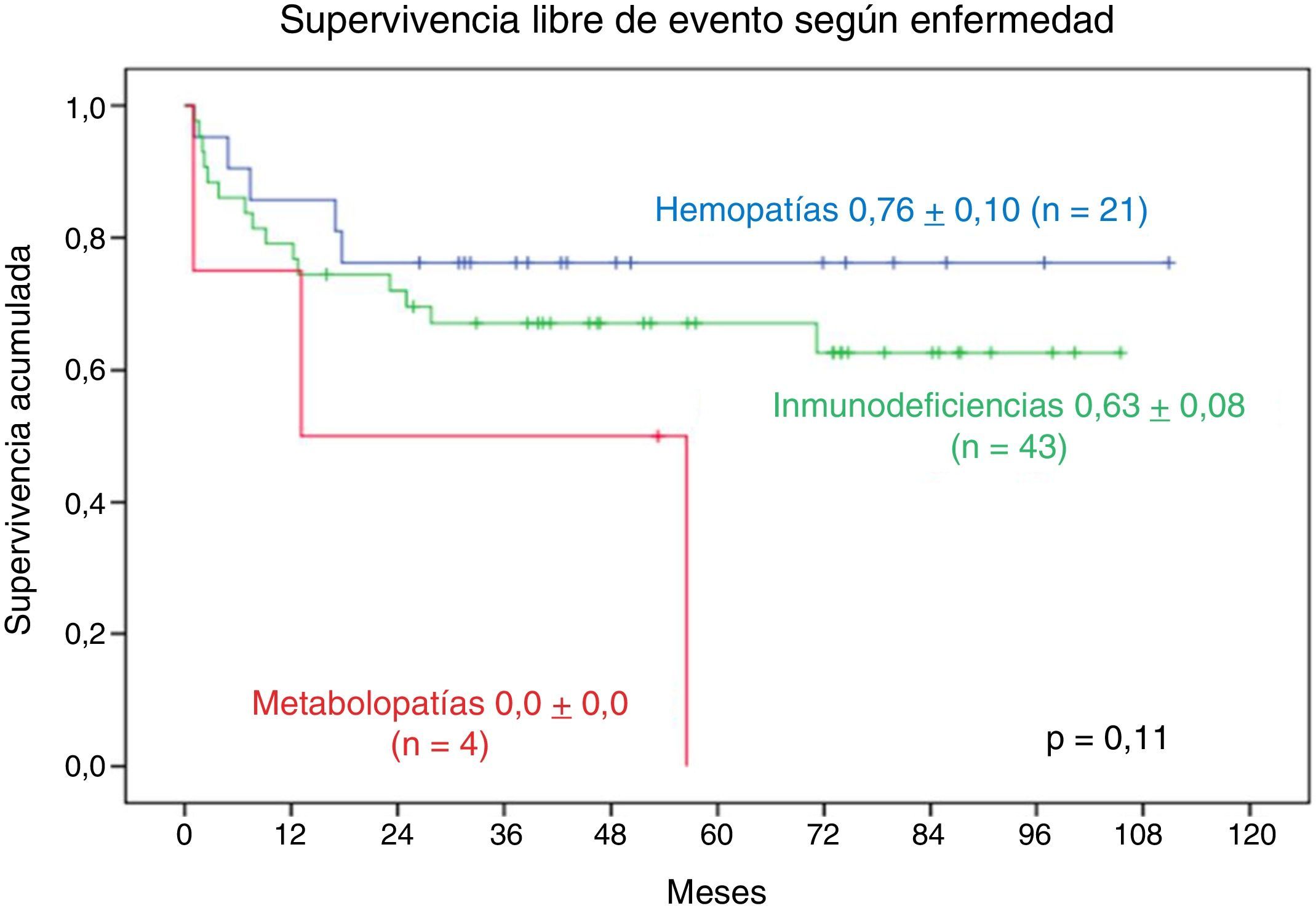
La SLE a 9 años en los pacientes con hemopatía es de 0,76±0,10, en los pacientes con IDP es de 0,63±0,08. La SLE a 2 años de seguimiento en los pacientes con metabolopatía es de 0,0±0,0. No se observa diferencia significativa (fig. 6).
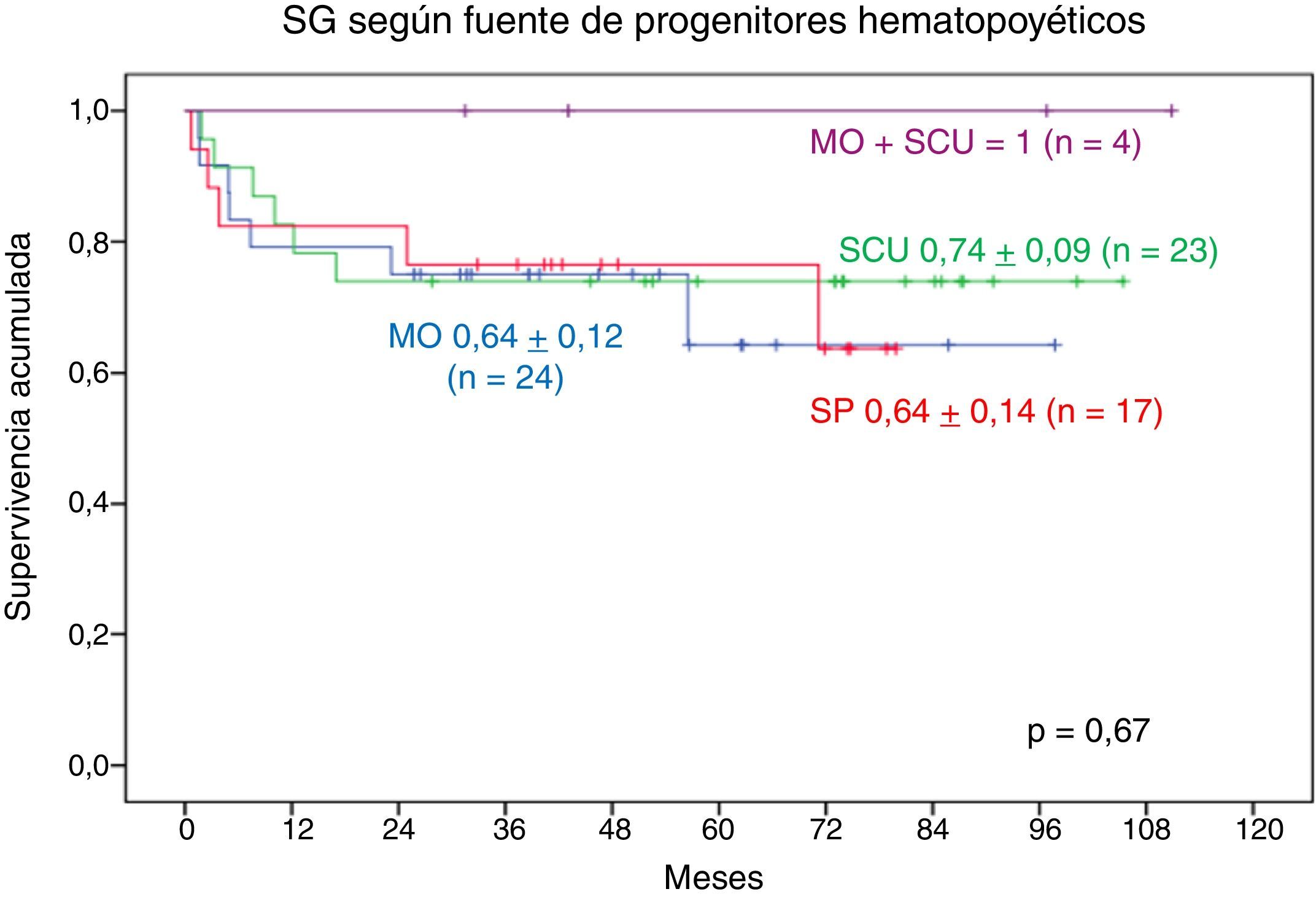
Según la fuente de los PH, la SG a 9 años en los pacientes trasplantados con SCU+MO es de 1, la SG a 9 años en los que recibieron SCU es de 0,70±0,11, La SG a 8 años en los pacientes trasplantados con MO es de 0,70±0,10 y la SG a 7 años en los pacientes trasplantados con SP es de 0,74±0,13. No se observa diferencia estadística significativa (fig. 7).
Según la familiaridad del donante de PH e identidad del HLA, la SG a 9 años en los pacientes trasplantados de DnE y HLA idéntico es de 0,77±0,11, en los pacientes trasplantados de DnE y HLA no idéntico es de 0,72±0,1. En cuanto a los donantes emparentados, la SG a 9 años con HLA idéntico es de 0,85±0,10 y a 7 años con HLA no idéntico es de 0,33±0,18. No se observa diferencia significativa (fig. 8).
Discusión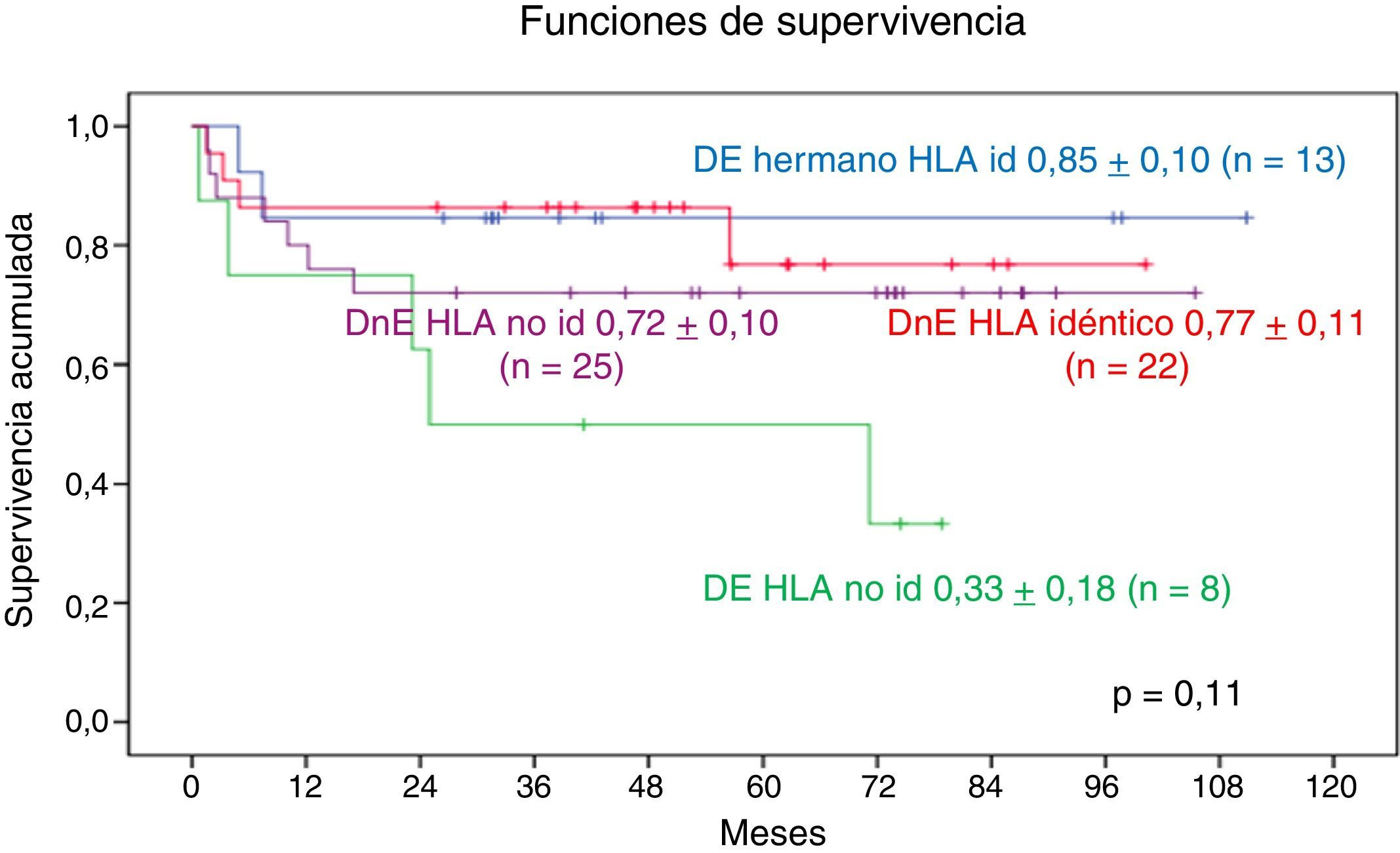
Este estudio expone los resultados obtenidos en 68 pacientes pediátricos con enfermedades genéticas sometidos a TPH con régimen de acondicionamiento de intensidad reducida recogidos por el GETMON en niños. Se ha conseguido reunir un número considerable de pacientes teniendo en cuenta que se estudian enfermedades genéticas con baja prevalencia en la población infantil.
Los pacientes con enfermedad genética tienen una mediana de edad al diagnóstico de 21 meses y una mediana de edad al trasplante de 46 meses. Esto nos indica que estos pacientes se trasplantan en los primeros años de vida, una vez diagnosticada la enfermedad y tan pronto se encuentra un donante de PH para intentar su curación.
En cuanto a la infusión de los PH, es destacable la inferioridad de CN y de CD34+ infundidas con la SCU, pero cabe destacar que los PH de la SCU presentan mayor potencial de multiplicación y formación de colonias que los obtenidos con MO o SP, lo cual compensa su menor número en términos absolutos. Cuando el donante de SCU es un hermano HLA idéntico, se puede recoger además MO para conseguir una celularidad total de progenitores más óptima, lo que en nuestro estudio nos ha aportado excelentes resultados.
El implante de neutrófilos del TPH con RIR ha sido superior al 90% en los 3 grupos de pacientes.
La experiencia en la literatura del TPH pediátrico con RIR es escasa. La mayoría de los estudios publicados hasta el 2007 muestran la experiencia de un único centro con un pequeño grupo heterogéneo de pacientes con regímenes de acondicionamiento variables y poco tiempo de seguimiento.
Bacigalupo et al.12 publicaron un trabajo con 35 pacientes alotrasplantados en Jerusalén. Los pacientes presentaban enfermedades no malignas y autoinmunes. Como acondicionamiento, se utilizó en todos ellos Flu+busulfán+GAT. La SG fue del 95% y la SLE fue del 80% a 2 años de seguimiento.
Shenoy et al.13 presentan un estudio de 16 pacientes tratados con Flu+melfalán+alemtuzumab. Los pacientes estaban diagnosticados de diferentes enfermedades no malignas y autoinmunes. Recibieron como fuente de progenitores MO o SP o SCU, tanto de donantes emparentados como de DnE. En sus resultados, todos los pacientes supervivientes (75%) se encuentran bien, con reversión o mejoría de su enfermedad de base.
Según la fuente de PH, los mejores resultados respecto a la SG se han obtenido con la combinación de SCU+MO de hermano HLA idéntico, con una SG a 9 años de 1. Parikh et al.14 publicaron en 2014 un estudio con 22 pacientes diagnosticados de enfermedades genéticas y trasplantados de SCU de DnE, siendo además la mayoría de ellos HLA no idéntico. El protocolo de RIR consistió en Flu+melfalán+tiotepa y alemtuzumab. En sus resultados, la SG a los 31 meses de seguimiento fue del 68,2%. En nuestra serie, la SG a 9 años en los pacientes trasplantados de SCU de DnE ha resultado del 70%, presentando mejores resultados que en el estudio de Parikh et al.
Respecto a la familiaridad e identidad del HLA, los mejores resultados en cuanto a la SG se han obtenido, como cabe esperar en enfermedad genética, cuando el donante es emparentado y con HLA idéntico.
En cuanto al TPH en los pacientes afectados de talasemia mayor, Lisni et al.15 han publicado recientemente un interesante estudio en el que presentan 60 pacientes. Acondicionamiento RIR: Tre+Flu+TT. Veinte pacientes se trasplantan de hermano HLA idéntico y 40 de DnE. La SG y la SLE a 5 años es del 93 y el 84%, respectivamente. Así como en nuestro estudio, siguiendo el protocolo del grupo italiano, la preparación basada en Tre ha demostrado ser segura y efectiva para pacientes con talasemia que reciben un alo-TPH, con una SG y SLE del 100%.
En la anemia de Fanconi el régimen con ciclofosfamida+Fluha demostrado ser seguro y efectivo. Se recomienda el uso de RIR en estos pacientes debido a la particular sensibilidad que presentan a los agentes tóxicos.
El grupo del Great Ormond Street Hospital8 publicó una serie de 113 pacientes con IDP que habían recibido un alo-TPH con RIR entre los años 1998 y 2006. La mayoría recibió un régimen que consistía en Flu+melfalán+alemtuzumab. La SG fue del 82%, con una mediana de seguimiento de 2,9 años. En nuestro estudio, la SG utilizando un régimen de acondicionamiento similar ha resultado del 70%, con una mediana de seguimiento de 5,2 años.
En las metabolopatías se han detectado situaciones de quimerismo mixto y fallo del implante, tal como hemos observado en nuestra pequeña serie. El TPH en esta enfermedad requiere una consideración individualizada para sopesar en cada paciente los riesgos y los beneficios que comporta el acondicionamiento con intensidad reducida, ya que este presenta una experiencia limitada y merece de mayor investigación16,17.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
