
El gen TSC2, responsable de la esclerosis tuberosa, se encuentra en el cromosoma 16p13.3, adyacente al gen de la poliquistosis renal autosómica dominante PKD1. Una deleción de gran tamaño puede afectar a ambos genes produciendo el llamado «síndrome de deleción de genes contiguos TSC2/PKD1» (MIM#600273). Se caracteriza por la presencia de quistes renales congénitos o de aparición muy precoz, en pacientes con esclerosis tuberosa, e implica un peor pronóstico de la enfermedad renal. Presentamos el caso de un niño de 6 años con esclerosis tuberosa, que en el período neonatal presentaba múltiples quistes renales de gran tamaño y bilaterales, realizándose posteriormente un estudio de confirmación genética mediante la técnica MLPA.
The TSC2 gene responsible for Tuberous Sclerosis, is located in chromosome 16p 13.3, adjacent to the gene for autosomal dominant polycystic kidney disease. A large deletion can involve both genes, causing the so-called TSC2/PKD1 contiguous gene syndrome (MIM#600273). It is characterized by congenital renal cysts, or their early onset in patients with tuberous sclerosis, and implies a worst prognosis in renal disease. We report the case of a five year-old boy with tuberous sclerosis, who presented with multiple large bilateral renal cysts in the neonatal period. A genetic confirmation study was later performed using the multiple ligation probe amplification (MLPA) technique.
La esclerosis tuberosa es una enfermedad de herencia autosómica dominante, caracterizada por la presencia de múltiples hamartomas a nivel del cerebro, la piel, los ojos y los riñones1-4. Su incidencia es de uno entre 6.000 y 11.000 nacidos vivos. Se debe tanto a mutaciones en TSC1 como en TSC2, genes supresores tumorales que codifican respectivamente la hamartina y la tuberina. Ambas proteínas están implicadas en la regulación de la diferenciación y proliferación celular. El gen TSC2 está situado en el cromosoma 16p13.3 adyacente al gen PKD1, responsable de la mayoría de los casos de poliquistosis renal autosómica dominante (PQRAD)1-10. Una deleción de gran tamaño puede afectar a los 2 genes, produciendo el llamado «síndrome de deleción de genes contiguos TSC2/PKD1»1,2,8,9,11,12, descrito por primera vez por Brook-Carter et al. en el año 19949. Este síndrome se caracteriza por la presencia de quistes renales bilaterales de gran tamaño, congénitos o de aparición muy precoz, en pacientes con esclerosis tuberosa, lo que modifica el pronóstico de la enfermedad renal.
Presentamos el caso de un varón de 6 años de edad con esclerosis tuberosa y quistes renales bilaterales objetivados en el período neonatal. Se realiza un estudio de confirmación genética mediante la técnica MLPA con el diagnóstico de síndrome de deleción de genes contiguos TSC2/PKD1.
Caso clínicoVarón de 6 años de edad, sin antecedentes personales ni familiares de interés, segundo hijo de una pareja joven. Madre de 33 años y padre de 35 años, sin historia de consanguinidad, con una hermana de 7 años también sana. En el tercer trimestre de gestación se realizó diagnóstico intraútero de tumoraciones cardíacas. El parto fue eutócico, a las 35 semanas, con exploración normal al nacimiento. Ante la sospecha de esclerosis tuberosa, en el período neonatal se realizaron diversas pruebas: ecografía cardíaca, con 3 rabdomiomas sin existir compromiso hemodinámico; RM cerebral, con nódulos subependimarios y tuberosidades corticales, y ecografía abdominal, con riñones aumentados de tamaño (50mm el derecho y 56mm el izquierdo), así como múltiples quistes renales bilaterales, sobre todo corticales, con pobre diferenciación córtico-medular. En la valoración oftalmológica se evidenciaron hamartomas retinianos. Ante estos hallazgos, se diagnosticó de esclerosis tuberosa sobre la base de criterios clínicos. A los 4 meses de edad, aparecieron 2 manchas cutáneas hipocrómicas y el paciente desarrolló hipertensión arterial, con cifras en torno a 130/80mmHg (siempre por encima del percentil 95). Se inició entonces tratamiento con amlodipino 5mg/24h. Inicialmente, evolucionó con un leve retraso madurativo, adquiriendo la marcha con 24 meses.
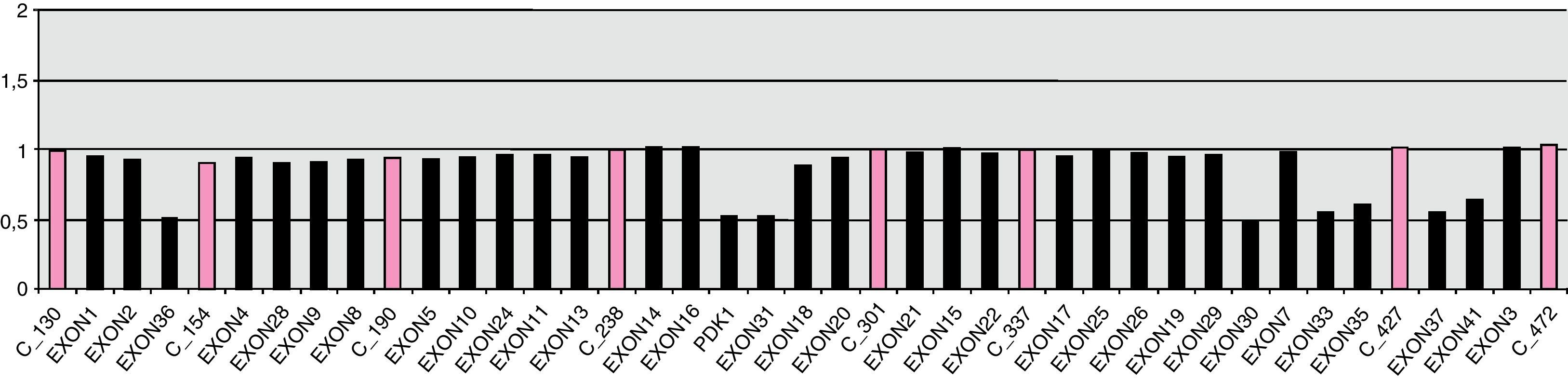
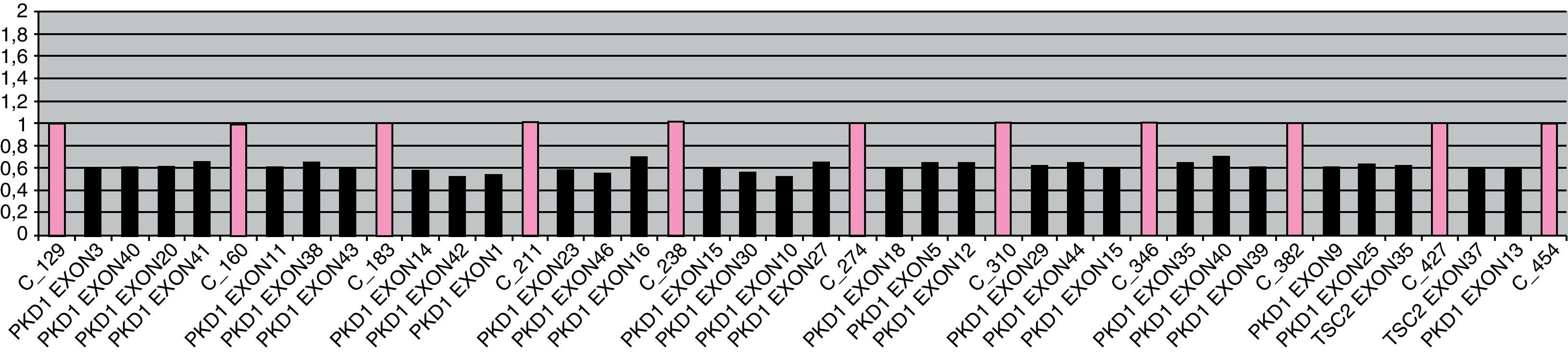
Con 3 años presentaba un incipiente angiofibroma facial y necesitaba 2 fármacos (amlodipino 5mg/24h y enalapril 5mg/12h) para el control de sus cifras tensionales. A esa edad, se solicitó estudio de genético de esclerosis tuberosa para consejo genético. El estudio molecular mediante MLPA mostró una deleción de los exones 30 al 41 del gen TSC2 en heterocigosis (fig. 1), así como de una copia completa del gen PKD1 (fig. 2). Ni los padres ni la hermana mostraron esta alteración a nivel periférico, sin poder descartar la existencia de un mosaicismo germinal. Con estos hallazgos, se diagnosticó de síndrome de deleción de genes contiguos TSC2/PKD1.
.")
.")
Con 6 años, el niño presenta angiofibromas faciales y múltiples manchas hipocromas en el tórax y el abdomen. En la exploración abdominal se palpan masas renales. Continúa recibiendo tratamiento con 2 fármacos antihipertensivos, estando preservada la función renal. Hasta ahora no ha presentado crisis epilépticas. Se le ha realizado una evaluación psicopedagógica que muestra una capacidad cognitiva dentro de la media. Además, cumple criterios clínicos de trastorno por déficit de atención con hiperactividad, que se maneja con medidas no farmacológicas.
DiscusiónLa esclerosis tuberosa en una enfermedad multisistémica genéticamente determinada, debida, en 2 tercios de los pacientes, a una mutación de novo2,3. El fenotipo es más leve si la mutación afecta al gen TSC1, tanto en los casos esporádicos como en los familiares2,5.
Las complicaciones renales son una manifestación frecuente de la esclerosis tuberosa y suponen la segunda causa de muerte tras la afección neurológica3,5,6. Los angiomiolipomas son la manifestación renal más frecuente1,3, encontrándose hasta en el 70-90% de los pacientes adultos. En niños, la frecuencia es menor, pero están presentes hasta en el 16% en menores de 2 años1. También pueden encontrarse quistes y carcinoma de células renales. Los quistes suelen ser asintomáticos y de pequeño tamaño3. Su frecuencia de aparición es similar en los casos secundarios a mutación en el gen TSC1 y en el TSC2, aunque se ha objetivado que los quistes aparecen de forma más precoz y son de mayor grado (de 2 a 4) con mutaciones en TSC25.
La PQRAD es una enfermedad de herencia autosómica dominante con una incidencia de uno entre 1.000 nacidos vivos3,10. Se produce en el 85% de los casos por mutación en el gen PKD1 y en un 15% por mutación a nivel del gen PKD27. Se caracteriza por la aparición, en la edad adulta, de quistes renales bilaterales que progresivamente conducen a un fallo renal. En ocasiones se acompaña de manifestaciones extrarrenales, como quistes hepáticos o pancreáticos, y aneurismas intracraneales3.
Se estima que hasta un 5% de los pacientes con esclerosis tuberosa presentan poliquistosis renal y viceversa12. Se ha demostrado que tanto la técnica MLPA como la del array -CGH, son métodos fiables para la detección de grandes deleciones, como la descrita previamente8,11. Brook-Carter et al. son los primeros en describir este síndrome en el año 19949. Encuentran grandes deleciones en el cromosoma 16, afectando a ambos genes (TSC2/PKD1), en 6 pacientes con esclerosis tuberosa y presentación en la infancia de poliquistosis renal9. Años más tarde, Sampson et al. realizan un estudio en el que encuentran esta deleción en 22 de los 27 pacientes estudiados con PQRAD y esclerosis tuberosa6.
Ambas enfermedades (esclerosis tuberosa y PQRAD) cursan con quistes renales, por lo que es difícil definir la contribución de cada gen al síndrome de deleción de genes contiguos TSC2/PKD13. Todavía no se conoce el mecanismo fisiopatológico que justifique la aparición precoz de la poliquistosis renal en este síndrome, aunque podría estar en relación con la inactivación completa del gen PKD13,6. Por el contrario, en la PQRAD, se preserva parte de la actividad de este gen, lo que podría justificar la presentación más tardía y menos severa de la enfermedad, ya en la edad adulta, así como algunas diferencias a nivel histopatológico6. La serie de Sampson et al. destaca que los pacientes con deleción TSC2/PKD1 presentan fallo renal antes que aquellos con mutación aislada del gen PKD16. De los 17 pacientes portadores de esta deleción, en 14 se detectaron quistes renales en los primeros 8 meses de vida, con características radiológicas similares a las encontradas en la PQRAD avanzada. Doce precisaron tratamiento con fármacos antihipertensivos, presentando la mayoría de ellos disminución en la tasa de filtrado glomerular, con insuficiencia renal terminal en 3 casos en torno a los 20 años. En esta serie, los mosaicismos se caracterizaron por un fenotipo más leve, detectándose los quistes de forma más tardía (entre los 3 y 35 años de edad), con características radiológicas similares a las encontradas en los previos, pero sin verse afectada la función renal6.
La combinación de alteraciones genéticas en TSC2 y PKD1 condicionaría, por lo tanto, una afectación renal más grave. En el artículo de Bonnet et al., mediante modelos de ratón, se ha evidenciado que TSC1, TSC2 y PKD1 interaccionan controlando la longitud de los cilios a nivel renal. Una mala orientación de los mismos, por cambios en su polaridad, podría ser un paso importante en la cistogénesis13.
Nuestro paciente ha presentado un desarrollo madurativo prácticamente normal y libre de crisis. Es probable que en el futuro llegue a desarrollar un fracaso renal crónico, dado el peor pronóstico renal que implica. Sin embargo, esta no se asocia a un peor pronóstico en lo referente a los quistes hepáticos y pancreáticos, o a los aneurismas intracraneales8, aunque estos últimos podrían ejercer un efecto sumatorio en las complicaciones neurológicas intrínsecas que conlleva la esclerosis tuberosa.
Por lo tanto, se debe sospechar un síndrome de deleción de genes contiguos (TS2/PKD1) en aquellos pacientes con esclerosis tuberosa que presenten, de forma congénita o en la infancia, quistes renales bilaterales, sin historia familiar previa, ya que modifica el pronóstico de la enfermedad renal.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

