






El síndrome hemofagocítico (SHF) o linfohistiocitosis hemofagocítica es una entidad con elevada mortalidad, típicamente reconocida en la edad pediátrica. Sin un correcto tratamiento, el SHF puede ser fatal: el riesgo de una rápida progresión a fallo multiorgánico y de afectación del sistema nervioso central con secuelas a largo plazo, son las consecuencias más graves de un retraso diagnóstico. Por lo tanto, el SHF es una urgencia médica que los pediatras deben saber identificar en un paciente con fiebre y afectación progresiva del estado general. La aplicación de los criterios diagnósticos de SHF, que consideran datos clínicos y analíticos (incluyendo un aspirado de médula ósea), y la búsqueda del factor desencadenante (infeccioso, oncológico, reumatológico, metabólico), son claves para poder instaurar un tratamiento dirigido, que neutralice el desencadenante y frene la hiperinflamación. En la presente revisión se exponen los datos más relevantes sobre la fisiopatología, diagnóstico y tratamiento de esta grave enfermedad para pediatras generales.
Haemophagocytic syndrome, or haemophagocytic lymphohistiocytosis (HLH), is a disorder with high mortality, typically recognised at paediatric age. Without proper treatment, HLH can be fatal. The risk of a rapid progression to multi-organ failure and central nervous system involvement leading to long-term sequelae, are the most feared consequences of a diagnostic delay. Therefore, HLH is a medical emergency that paediatricians should be able to identify in a patient with fever and progressive worsening of general condition. The application of the HLH diagnostic criteria, which include clinical and analytical data (as well as a bone marrow aspirate), and the search for a trigger (infectious, oncological, rheumatological, or metabolic). These are decisive for the establishment of a targeted treatment, which aims at neutralising the trigger and reducing the hyper-inflammation. The most relevant data for general paediatricians are presented in this review, including the physiopathology, diagnosis, and treatment of this serious disease.
El síndrome hemofagocítico (SHF), también conocido como linfohistiocitosis hemofagocítica (HLH), es un síndrome clínico provocado por una respuesta inadecuada del sistema inmunológico a un desencadenante, sea infeccioso, neoplásico, reumatológico o metabólico, dando lugar a una reacción inflamatoria exagerada. Esta respuesta inmune altamente activada causa una liberación exagerada o tormenta de citocinas responsable del cuadro clínico. SHF no es una única enfermedad, sino un síndrome clínico asociado a gran variedad de causas subyacentes que conducen al mismo fenotipo inflamatorio característico1,2.
El término hemofagocitosis describe los hallazgos característicos de macrófagos activados que incorporan eritrocitos, leucocitos, plaquetas y precursores. Puede encontrarse en otras situaciones clínicas, y su consecuencia es la destrucción (digestión) de células sanguíneas en médula ósea y otros tejidos. Cuando la hemofagocitosis ocurre en el contexto de una respuesta inmune altamente estimulada pero ineficaz, se denomina SHF. Se produce una infiltración multiorgánica de linfocitos y macrófagos activados: en caso de realizarse una biopsia de los órganos afectos, observaríamos fenómenos inflamatorios y hemofagocitosis.
SHF puede aparecer a cualquier edad, aunque la forma primaria suele ocurrir en la infancia. Se describió inicialmente asociado a infecciones virales, pero posteriormente se ha relacionado con otras infecciones por hongos, bacterias y parásitos (en particular, leishmaniasis)2,3. Las infecciones y especialmente el virus Epstein-Barr (VEB) es un desencadenante común, tanto en SHF primario como secundario. También puede ocurrir en el contexto de enfermedades autoinmunes como artritis idiopática juvenil, lupus eritematoso sistémico, o enfermedad de Kawasaki4. De hecho, cualquier estimulación intensa de la inmunidad celular (infección, reumatismo, tumor) podría desencadenar una forma secundaria de SHF. SHF asociado a enfermedades reumáticas se denomina síndrome de activación macrofágica.
Tanto la HLH primaria como secundaria pueden ser rápidamente fatales y requieren tratamiento precozmente5–7.
FisiopatologíaEl SHF es un cuadro inflamatorio grave causado por una proliferación y activación de linfocitos y macrófagos, que secretan grandes cantidades de citocinas. Dicho proceso a menudo es precipitado por un desencadenante infeccioso, que actúa sobre un sistema inmune disfuncional, ya sea por un defecto primario/genético, o secundario a diferentes enfermedades (autoinmunes, autoinflamatorias, oncológicas, metabólicas) o tratamientos inmunomoduladores. Diferentes trabajos en modelos murinos8 han demostrado que el defecto de la función citotóxica de linfocitos CD8+ es crucial en la fisiopatología. De forma fisiológica, la función citotóxica de las células CD8+ y NK permite la lisis de células afectadas (infectadas, neoplásicas, envejecidas, sobrantes…) a través de la exocitosis de sus gránulos citotóxicos (o degranulación), que contienen proteínas citolíticas (granzimas) y perforinas (proteínas que formarán un poro en la membrana plasmática de la célula diana, por donde entrarán las granzimas para su lisis). La retirada deficiente de las células afectadas estimula la presentación antigénica por parte de células dendríticas y se perpetúa la activación linfocitaria-T. La activación sostenida e incontrolada de estos linfocitos, que producen grandes cantidades de interferón-gamma (IFN-γ), tiene como consecuencia la activación macrofágica, que a su vez producirán hemofagocitosis y liberarán citocinas inflamatorias (TNFa, IFN-α, IL-6, IL-18, IL-12) que perpetúan la presentación antigénica y activación T, contribuyendo todo ello a las manifestaciones clínicas (fig. 1, tabla 1). Se produce un círculo vicioso inflamatorio y de citocinas liberadas. Dada la disfunción de la citotoxicidad, la función citotóxica suele evaluarse para el diagnóstico y suele estar disminuida o ausente, tanto en formas primarias como secundarias. La diferencia es que en las formas primarias, el defecto de citotoxicidad persiste en el tiempo incluso sin síntomas clínicos de SHF. La severidad se ha correlacionado con la función citotóxica residual: por ello, la intensidad de los síntomas puede variar según el defecto genético, tipo de mutación, y agente secundario desencadenante. Por último, en las formas de SHF secundario, la fisiopatología no está bien definida. Se sabe que no todo agente infeccioso puede desencadenar un SHF: dicho agente debe poseer unas características especiales. Destaca el VEB que puede interferir con la función de las células T-CD8+ a través de proteínas específicas y se producen altos niveles de citocinas, fundamentalmente IL-18, IFN γ 1,2.

Manifestaciones clínicas y de laboratorio. Mecanismos productores
| Manifestaciones cardinales | |
|---|---|
| Manifestaciones | Mecanismo productor |
| Fiebre | Elevación de IL-1 e IL-6 |
| Pancitopenia | Elevación de TNFa y hemofagocitosis |
| Hipertrigliceridemia | TNFa inhibe la lipoproteinlipasa |
| Hiperferritininemia | La ferritina es secretada por los macrófagos activados por el IFN gamma |
| Hipofibrinogenemia | El activador del plasminógeno es secretado por los macrófagos activados y causa una hiperfibrinólisis |
| Niveles altos de CD25s (receptor soluble de interleucina-2) | Los linfocitos activados son la fuente de CD25s |
| Hepatoesplenomegalia | Infiltración multiorgánica por linfocitos y macrófagos (histiocitos) activados |
| Otras manifestaciones |
|---|
| Clínicas: linfadenopatías, disfunción hepática, diátesis hemorrágica, edema, síntomas neurológicos, fallo multiorgánico |
| De laboratorio: leucopenia (linfopenia), hipoproteinemia, hipoalbuminemia, hiponatremia, hipertransaminasemia, hiperbilirrubinemia, elevación de LDH, elevación de D-dímeros |
Adaptado de Brisse et al.8.
Los signos clínicos iniciales del SHF son los mismos que los de cualquier proceso infeccioso grave. Se caracteriza por fiebre alta prolongada, que asocia de forma progresiva pancitopenia y hepatoesplenomegalia, junto con otros datos de fallo multiorgánico como afectación hepática, pulmonar, sistema nervioso central, etc.6,7. En ocasiones observamos síndrome de pérdida capilar e hipoalbuminemia. Como hallazgos de laboratorio podemos encontrar citopenias, coagulopatía, hipertrigliceridemia, hipofibrinogenemia, hipertransaminasemia, e hiperferritinemia7 (tabla 1). Estos eventos conforman los criterios clínicos y de laboratorio utilizados para el diagnóstico, establecidos en 1991 y revisados en 20042,4,6 (tabla 2). El aspirado de médula ósea revela normalmente una maduración celular normal con hipercelularidad.
Criterios clínicos diagnósticos del síndrome hemofagocítico (protocolo HLH 2004)
| SHF familiar o de causa genética identificada |
| Criterios clínicos y de laboratorio (deben cumplirse 5 de 8) |
| 1. Fiebre ≥ 38,5°C |
| 2. Esplenomegalia |
| 3. Citopenias |
| --Hemoglobina <90 g/l (si menor de 4 semanas de vida, <120 g/l). |
| --Plaquetas < 100.000/mm3 |
| --Neutrófilos <1000 / mm3 |
| 4. Hipertrigliceridemia y/o hipofibrinogenemia |
| --Triglicéridos en ayunas ≥ 3 mmol/l |
| --Fibrinógeno <1,5 g/l |
| 5. Ferritina >500μg/l |
| 6. sCD25 ≥ 2400 U/ml |
| 7. Descenso o ausencia de actividad citotóxica NK |
| 8. Hemofagocitosis en médula ósea, LCR o ganglios linfáticos |
Fuente: Henter et al.6.
El SHF puede presentarse con síntomas neurológicos debido a la infiltración del SNC por macrófagos activados. La clínica es similar a la encefalitis. En LCR encontraremos pleocitosis de mononucleares (meningitis linfocitaria) o disociación albúmino-citológica. La afectación del SNC empeora de forma significativa el pronóstico y puede producir secuelas permanentes.
No todos los criterios diagno¿sticos se encuentran presentes inicialmente o en la presentación neonatal. La hemofagocitosis no es necesaria para el diagnóstico. Es la progresión de la presentación de los criterios lo que debe alertar al médico de un posible SHF1. La dificultad del diagnóstico reside en que es difícil distinguir entre la activación macrofágica fisiológica que puede observarse en sepsis9, enfermedades malignas o autoinmunes/autoinflamatorias, y la activación patológica que define SHF10.
En resumen, la mala o inusual progresión de los síntomas de una enfermedad común debe orientar la sospecha de SHF. Aunque los hallazgos microbiológicos confirmen una infección, esta puede ser la desencadenante del SHF que causa un fallo multiorgánico progresivo, que no responde al tratamiento antimicrobiano habitual y que precisa una terapia específica. Por ello, un diagnóstico y tratamiento precoces podrán mejorar el pronóstico de los pacientes.
Diagnóstico y diagnóstico diferencialDe acuerdo con los criterios clínicos y analíticos propuestos por la Histiocyte Society6 (tabla 2), el diagnóstico puede establecerse cuando se cumplen 5 o más de los 8 criterios clínicos y de laboratorio o cuando se confirma el diagnóstico molecular. Los estudios inmunológicos y genéticos son importantes, especialmente para diagnosticar las formas familiares o primarias.
Generalmente en niños fuera del periodo neonatal, la fiebre y esplenomegalia están presentes en el 90-100% de los pacientes1,4. La hiperferritinemia es un marcador muy importante, especialmente niveles muy elevados (ferritina > 10.000 ug/l en niños, 90% de sensibilidad y 96% de especificidad)4,7. La presencia de hemofagocitosis en médula ósea al inicio se observa solamente en 30-40% de los casos, dificultando el diagnóstico, y aumenta al 80-90% con la progresión de la enfermedad1. La cuantificación sérica del receptor de interleucina-2-soluble (rIL2s o CD25s) está incluido en los criterios (niveles > 2400 U/mL), por ser marcador de activación linfocitaria; estudios recientes muestran que el ratio de los niveles séricos de rIL2s y ferritina son útiles para diferenciar algunas formas de SHF11. El estudio de la actividad NK es la técnica más transversal a la mayoría de los defectos debido a la alteración de la capacidad de citotoxicidad característica. Desafortunadamente existen dificultades para solicitar la determinación de actividad NK y cuantificación sérica del CD25s, ya que solo están disponibles en laboratorios de inmunología especializados.
Uno de los mayores retos del HLH es su diagnóstico, dado que los signos y síntomas iniciales son poco específicos (fig. 2). Su espectro clínico obliga a incluirlo en el diagnóstico diferencial de: fiebre de origen desconocido; fallo hepático agudo o hepatitis con coagulopatía (30% presentan aumento de transaminasas >100 U/l)12; sepsis con fallo multiorgánico; meningitis o encefalitis linfocitaria con lesiones focales en SNC; en el periodo neonatal, formas aisladas de afectación SNC o insuficiencia hepática fulminante, que simulan enfermedad metabólica.

Algoritmo diagnóstico. Esquema adaptado de Cincinnati Children's HLH Diagnostic Strategy en: www.cincinnatichildrens.org/dchi
El estudio por citometría de flujo de proteínas implicadas en la fisiopatología puede ser de gran utilidad, como en la deficiencia de perforina (FLH-2), SAP (XLP-1) o XIAP (XLP-2) (fig. 2), aunque todavía no son criterios incluidos en el protocolo HLH-20046.
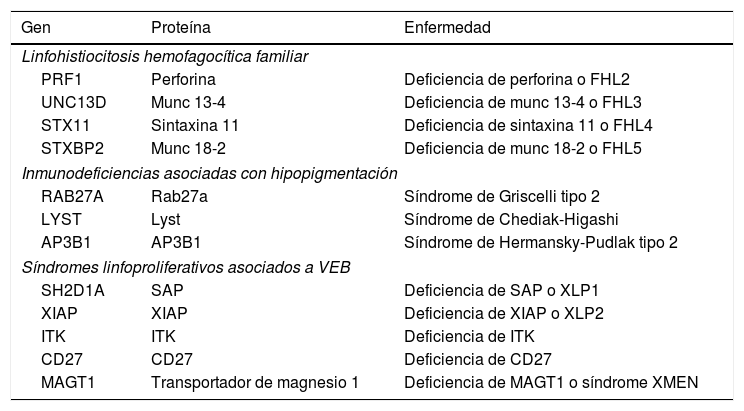
En los últimos años, se han identificado defectos genéticos que afectan a la citotoxicidad linfocitaria y que son la principal causa de SHF primario1,4,13,14. Pero todavía existen casos (hasta el 30%) con sospecha de primario (incluso con historia familiar o consanguinidad) donde no se identifica la alteración molecular15 y se estima que hay otros genes implicados todavía desconocidos. Recientemente, se han descrito casos con mutaciones en heterocigosis o combinadas entre genes de FHL14.
Las formas primarias incluyen linfohistiocitosis hemofagocítica familiar (FHL), inmunodeficiencias asociadas con albinismo óculo-cutáneo y síndromes linfoproliferativos asociados al VEB (tabla 3). Se han descrito mutaciones en 12 genes diferentes con patrón de herencia autosómica recesiva o ligada al cromosoma X. Los pacientes con SHF presentan heterogeneidad genética, pero suelen compartir un inmunofenotipo común por el defecto en la citotoxicidad13,14.
Formas genéticas de síndrome hemofagocítico
| Gen | Proteína | Enfermedad |
|---|---|---|
| Linfohistiocitosis hemofagocítica familiar | ||
| PRF1 | Perforina | Deficiencia de perforina o FHL2 |
| UNC13D | Munc 13-4 | Deficiencia de munc 13-4 o FHL3 |
| STX11 | Sintaxina 11 | Deficiencia de sintaxina 11 o FHL4 |
| STXBP2 | Munc 18-2 | Deficiencia de munc 18-2 o FHL5 |
| Inmunodeficiencias asociadas con hipopigmentación | ||
| RAB27A | Rab27a | Síndrome de Griscelli tipo 2 |
| LYST | Lyst | Síndrome de Chediak-Higashi |
| AP3B1 | AP3B1 | Síndrome de Hermansky-Pudlak tipo 2 |
| Síndromes linfoproliferativos asociados a VEB | ||
| SH2D1A | SAP | Deficiencia de SAP o XLP1 |
| XIAP | XIAP | Deficiencia de XIAP o XLP2 |
| ITK | ITK | Deficiencia de ITK |
| CD27 | CD27 | Deficiencia de CD27 |
| MAGT1 | Transportador de magnesio 1 | Deficiencia de MAGT1 o síndrome XMEN |
Las mutaciones en el gen PRF1 (FHL2) que codifica para la proteína perforina, fue el primer defecto genético asociado. La deficiencia de perforina representa aproximadamente el 35%15 de las alteraciones moleculares, aunque varía según poblaciones. El cribado de perforina por citometría de flujo es altamente sensible y específico.
Los defectos genéticos en UNC13D causan el tipo FHL3 y produce la deficiencia de Munc 13-4 (35% de FHL). En el sur de Europa, FHL2 y FHL3 juntos representan casi el 70%15. El defecto de sintaxina 11 o FHL4 solo afecta al 5%, y finalmente la deficiencia de Munc 18-2 o FHL5 afecta aproximadamente al 5-20%. Los defectos FHL3, 4 y 5 tienen severamente alterada la capacidad citotóxica NK y por tanto el estudio de actividad NK es obligado12,16.
Inmunodeficiencias asociadas a albinismo óculo-cutáneoLos pacientes con síndrome de Griscelli-tipo 2, síndrome de Chediak-Higashi o síndrome de Hermansky-Pudlak-tipo-2 (HP2) presentan defectos del tráfico y exocitosis de los lisosomas secretores que tienen funciones importantes en numerosos tipos celulares, que provocan enfermedades neurológicas progresivas, hipopigmentación o albinismo, coagulopatía, neutropenia y alteración de la actividad NK. Los pacientes habitualmente presentan pelo de color plateado o blanco-grisáceo, cuyo análisis al microscopio óptico puede ayudar al diagnóstico. La presencia de neutrófilos vacuolados en sangre periférica puede orientar la sospecha de Chediak-Higashi. La queratosis palmoplantar orienta a HP2. Este tipo de inmunodeficiencias primarias pueden presentarse como SHF17.
Síndromes linfoproliferativos asociados al virus de Epstein-BarrEn varones se ha encontrado una fuerte asociación entre SHF desencadenado por VEB y síndromes linfoproliferativos ligados al cromosoma X (XLP1, XLP2, cuyas proteínas, SAP (XLP1) o XIAP (XLP2) pueden evaluarse por citometría de flujo, y deficiencia de MAGT1)18. También se han descritos defectos ITK y CD27 con herencia autosómica recesiva. Algunos casos pueden interpretarse inicialmente como mononucleosis infecciosa por VEB, pero evolucionan y se comportan como formas graves de SHF3.
Síndrome hemofagocítico adquirido o secundario (tabla 4)SHF secundario es clínicamente idéntico al SHF primario, pero no existe historia familiar o defecto genético relacionado con la citotoxicidad. Se han descrito numerosos casos fundamentalmente asociados a infecciones de cualquier tipo (virus, bacterias, hongos y protozoos), enfermedades autoinflamatorias o autoinmunes, malignas y metabólicas. Virtualmente toda infección no controlada puede presentar criterios clínicos compatibles con SHF. Destaca la infección por Leishmania que debe descartarse siempre, en sangre periférica o médula ósea (tinción o PCR).
Causas más comunes de síndrome hemofagocítico secundario o adquirido
| Infecciones | Neoplasias | Enfermedades reumatológicas | Enfermedades |
|---|---|---|---|
| Virus (VEB > VHS,CMV, hepatotropos, dengue, enterovirus, PVB19) | Linfoma | Artritis idiopática juvenil | Intolerancia lisinúrica |
| Bacterias y micobacterias | Leucemia | Lupus eritematoso sistémico | Biotinidasa |
| Hongos | Melanoma | Vasculitis (PAN) | Aciduria mevalónica |
| Parásitos (Leishmania) | Otros tumores | Autoinflamatorias (NLRC4) Enf. Kawasaki | Déficit de transaldolasa Enf. Gaucher Enf. Wolman |
En otros casos el SHF es secundario a otras inmunodeficiencias adquiridas por quimioterapia, inmunosupresión, terapias biológicas y trasplantes1,13. La asociación con neoplasias malignas puede aparecer a cualquier edad, de forma concomitante con el inicio y diagnóstico del cáncer o durante su tratamiento18.
Las enfermedades reumatológicas, fundamentalmente al comienzo o en caso de falta de control de la enfermedad, como artritis idiopática juvenil, lupus eritematoso sistémico o deficiencia de NLRC4, son causas frecuentes de SHF secundario. Suele denominarse síndrome de activación macrofágica9.
En la mayoría de los casos, el comienzo del SHF adquirido es más tardío que SHF primario, el agente desencadenante suele estar identificado y la recurrencia de la enfermedad es menor19.
Pruebas del laboratorio de inmunología (fig. 2)Las pruebas fundamentales del laboratorio de inmunología para el diagnóstico de SHF consisten en técnicas basadas en citometría de flujo:
- -
1. Tinción intracitoplasmática de perforina (cribado FHL2).
- -
2. Ensayo de degranulación de CD107a (cribado FHL3-5 y casos sin diagnóstico molecular).
- -
3. Ensayo de citotoxicidad NK (evalúa el defecto globalmente).
De estas pruebas, la combinación de las técnicas 1 + 2 es superior al ensayo clásico de función NK (menor sensibilidad y especificidad20). Los estudios inmunológicos han permitido establecer diferencias entre formas primarias y secundarias13. Se observan patrones diferentes de activación y diferenciación de linfocitos T y de los estudios de degranulación13,21. Sin embargo, los resultados no suelen estar disponibles de forma inmediata, por lo que las decisiones terapéuticas iniciales se deben tomar según la gravedad del cuadro clínico6.
TratamientoEl objetivo general es la supresión y control de la hiperinflamación e hipercitocinemia y la eliminación de células activadas e infectadas. En formas genéticas, el único tratamiento curativo es el trasplante de progenitores hematopoyéticos (TPH) para corregir el defecto de citotoxicidad. Las diferentes modalidades de tratamiento incluyen corticoides (primer escalón terapéutico, habitualmente dexametasona 10 mg/m2 según protocolos HLH-94, HLH-2004, en formas primarias, metilprednisolona en secundarias), inmunosupresores, citostáticos, inmunomoduladores, anticuerpos monoclonales y agentes anticitocinas (tabla 5)5,6.
El tratamiento debe ajustarse a la gravedad clínica y evolución. Hay formas muy graves de progresión fulminante que requieren terapia específica y medidas de soporte intensivo de forma empírica y urgente, mientras que otros casos son más leves o cursan de forma recidivante y responden a tratamientos menos agresivos3,5.
Las decisiones terapéuticas son complejas porque muchos pacientes presentan infecciones con gérmenes identificados y se prioriza el tratamiento antimicrobiano que resulta insuficiente para frenar la cascada inflamatoria. Aunque parece contradictorio, se necesitan administrar fármacos inmunosupresores como esteroides, bloqueantes de citocinas, gammaglobulinas o incluso citostáticos para destruir las células activadas. En formas graves los pacientes precisan ingreso en Unidades de Cuidados Intensivos Pediátricos porque requieren ventilación mecánica, soporte hemodinámico, transfusiones etc. Se aconseja consultar con expertos y trasladar a los pacientes graves a centros terciarios5.
La búsqueda exhaustiva del agente causante, identificación microbiológica y administración de terapia antibiótica, antifúngica, antiviral o antiparasitaria adecuada son fundamentales1,3,5,6. En la mayoría de los casos no es suficiente una correcta terapia antiinfecciosa y debe asociarse precozmente un tratamiento específico de SHF, excepto en leishmaniasis que suelen resolverse con terapia antimicrobina como anfotecina-B-liposomal.
En casos graves y progresivos, se recomienda empezar a tratar en base a una fuerte sospecha clínica, aunque no cumplan estrictamente todos los criterios diagnósticos, ya que es una situación urgente de riesgo vital. Las formas moderadas suelen responder bien a esteroides e inmunoglobulinas, pero los casos más graves deben tratarse con combinaciones de fármacos inmunosupresores o citotóxicos.
Sin tratamiento, el pronóstico de SHF es muy malo, especialmente en formas genéticas22. La introducción del etopósido en los años 90 representó un importante avance terapéutico. Destacan los resultados del protocolo HLH-9423 (Histiocyte Society) basado en la combinación de dexametasona, etopósido, ciclosporina-A y TPH en pacientes con formas familiares, recidivantes o severas y persistentes. Este estudio consiguió mejorar la supervivencia global, con tasas a 5 años del 54%, aunque el 29% fallecían antes del TPH. Los resultados a largo plazo se mantenían24, pero el 22% desarrollaban secuelas neurológicas25.
El siguiente estudio HLH-20047 demostró el beneficio de esteroides y etopósido en una cohorte de 369 niños de 27 países, incluyendo España26. Las modificaciones propuestas de administrar ciclosporina-A desde el inicio y recomendar TPH cuando hubiera un donante disponible, no consiguieron avances significativos en la supervivencia (62% a 5 años) ni en reducción de mortalidad precoz (19%). La supervivencia pos-TPH a 5 años fue similar (67% en HLH-2004, 66% en HLH-94)23,26. Se identificaron factores de mal pronóstico: afectación neurológica y pleocitosis en LCR, cifras elevadas de bilirrubina y ferritina al diagnóstico27. Desde 2012 no se han propuesto nuevos estudios colaborativos y en el momento actual se aconseja utilizar el protocolo HLH-94 con los criterios disgnósticos del HLH-2004.
Otros estudios han conseguido buenos resultados con gammaglobulina antitimocítica (ATG)28, alemtuzumab (anticuerpo anti-CD52)29 o rituximab (anticuerpo anti-CD20)30. Ensayos clínicos con anticuerpos monoclonales antiinterferón-gamma o híbridos de inmunoterapia (ATG, dexametasona y etopósido) todavía no han publicado los resultados. En formas refractarias se han observado respuestas a anakinra, alemtuzumab, tocilizumab, etanercept o ruxolitinib29.
La duración del tratamiento varía según la evolución y respuesta. En las formas primarias la terapia inicial es un puente hasta el trasplante: intenta conseguir la remisión y búsqueda del mejor donante disponible. En formas secundarias, la duración puede ser de pocos días o semanas si alcanza la remisión clínica, vigilando aparición de recaídas posteriores. En formas recidivantes puede ser necesario reiniciar el tratamiento específico y considerar el trasplante.
En las formas familiares o primarias, el TPH alogénico se considera la única terapia curativa para la reconstitución del sistema inmune deteriorado. También se recomienda en casos muy graves, progresivos y recidivantes o incluso en hermanos asintomáticos con defectos moleculares confirmados. Las complicaciones asociadas al TPH suelen ser importantes, por lo que se recomienda su realización en centros experimentados con utilización de protocolos de acondicionamiento de intensidad reducida25,31. Quimerismos parciales, estables y mixtos suelen ser suficientes31.
En SHF secundarias, las recomendaciones están menos establecidas, pero se deben evitar tratamientos prolongados. En formas graves asociadas a infecciones como VEB puede ser necesaria terapia intensiva incluso con etopósido o TPH3,5. El manejo de los SHF asociados a malignidad es muy complejo por la necesidad de combatir la neoplasia y SHF al mismo tiempo32. En síndromes de activación macrofágica33, como tratamiento de primera línea se propone los corticoides a dosis altas y ciclosporina, como segunda línea anakinra (inhibidor de interleukina-1) y excepcionalmente requieren etopósido.
ConclusionesEl SHF es una entidad con elevada mortalidad, típicamente reconocida en edad pediátrica.
Sin un tratamiento correcto, el SHF puede ser fatal: el riesgo de una rápida progresión a fallo multiorgánico y de afectación del sistema nervioso central con secuelas a largo plazo son las consecuencias más graves de un retraso diagnóstico.
Por lo tanto, el SHF es una urgencia médica que los pediatras deben saber identificar en un paciente con fiebre y afectación progresiva del estado general.
La aplicación de los criterios diagnósticos de SHF, que incluyen datos clínicos y analíticos, y la búsqueda del factor desencadenante (infeccioso, oncológico, reumatológico, metabólico), son claves para poder instaurar un tratamiento dirigido, que neutralice el desencadenante y frene la hiperinflamación.
FinanciaciónEste trabajo ha sido financiado en parte por el Fondo de Investigaciones Sanitarias (FIS) a favor de Luis M. Allende y Luis I. González-Granado (PI 16/2053). También por los fondos del Fondo de Investigaciones Sanitarias (FIS) a favor de Juana Gil Herrera e Itziar Astigarraga (PI12/02761) y por los fondos del Telemaratón de Enfermedades Raras de EITB/ BIOEF al proyecto de Histiocitosis de Itziar Astigarraga (BIO16/ER/020/BC). Cofinanciado con fondos FEDER.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los pacientes, familiares y profesionales que han participado en los estudios colaborativos. A Susana García-Obregón por su labor en los protocolos internacionales de histiocitosis en España.
Este artículo fue presentado previamente en el Congreso de la Asociación Española de Pediatría. Santiago de Compostela, 2 junio de 2017. Los autores participaron en la mesa redonda titulada «Síndromes hemofagocíticos. La importancia de un diagnóstico precoz».
