






El síndrome de Sotos se caracteriza por sobrecrecimiento con facies peculiar, macrocefalia, talla alta y alteraciones del desarrollo psicomotor. Presentamos a un paciente de 20 meses de edad con diagnóstico confirmado por genética molecular con detección de mutación nonsense en el gen NSD1 no descrita previamente, exhibiendo cutis laxa como la característica fenotípica más llamativa en el periodo neonatal. Esta asociación se había descrito previamente en 3 pacientes con diagnóstico clínico de síndrome de Sotos sin diagnóstico molecular confirmatorio. En nuestro paciente, la presencia de cutis laxa llevó al diagnóstico diferencial con los defectos congénitos de glucosilación. En el seguimiento posnatal presentó una somatometría con perímetro cefálico y talla mayores de p97 (cercano a p50 al nacimiento), junto con el desarrollo de rasgos fenotípicos característicos del síndrome de Sotos durante los primeros meses de vida, los que proporcionaron la clave para el diagnóstico clínico y la investigación molecular.
Sotos syndrome is an overgrowth condition characterized by facial gestalt, macrocephaly, excessive height, and different degrees of developmental delay. We report the case of a 20-month-old boy with a confirmatory molecular study, showing a novel nonsense mutation in NSD1 gene, presenting cutis laxa as the main phenotypic trait in the neonatal period. This association has been previously described in 3 patients with a clinical diagnosis of Sotos syndrome, without confirmatory molecular analysis. Our patient was tested for congenital disorders of glycosilation as part of the cutis laxa differential diagnosis. During the postnatal follow-up period the head circumference and height became greater than 97th percentile (having been close to the 50th in the newborn period). These facts and the progressive development of characteristic phenotypic features of Sotos syndrome during the first months of life gave us the clue for the clinical diagnosis and the molecular investigation.
El síndrome de Sotos (OMIM 117550), también conocido como gigantismo cerebral, es uno de los síndromes de sobrecrecimiento, junto principalmente con los síndromes de Wiedemann-Beckwith, de Weaver, de Simpson-Golabi-Behmel y de Bannayan-Riley-Ruvalcaba1. Los criterios diagnósticos han sido reevaluados en los últimos años, a consecuencia de la identificación de mutaciones en el gen NSD1 como causante del síndrome y de la revisión de las características fenotípicas de series amplias de estos pacientes con confirmación molecular2–4. Las manifestaciones clínicas incluyen tres hallazgos cardinales, presentes en el 90% de los pacientes: dismorfia facial característica, dificultad de aprendizaje y sobrecrecimiento (talla y/o perímetro cefálico > p98), 11 manifestaciones mayores, presentes en el 15% de los casos, y otras manifestaciones descritas en un número pequeño de pacientes4.
Describimos un paciente con diagnóstico genético confirmado a los 14 meses de vida, en el cual se identifica una nueva mutación nonsense en el gen NSD1. Asimismo, queremos destacar la peculiar forma de presentación clínica, siendo la cutis laxa la característica fenotípica más llamativa en el periodo neonatal.
Caso clínicoSe trata de una gestación controlada y normoevolutiva, con detección prenatal de ectasia piélica izquierda (9,7mm) e hidrocefalia leve (asimetría ventricular con ventrículo derecho mayor que el izquierdo). Amniocentesis con cariotipo masculino normal 46,XY. Parto eutócico a las 39 semanas, Apgar 9/10. Somatometría al nacer con peso ± desviación estándar de 3.340±0,05g, talla 51±0,58cm y perímetro cefálico 36±1,09cm, destacando únicamente escafocefalia en la exploración inicial.
A los 8 días de vida consulta el servicio de urgencias por hipoactividad, rechazo del alimento y pérdida de peso. En el examen inicial se detecta pérdida ponderal del 12,5% del peso al nacer, deshidratación isonatrémica, acidosis metabólica y uroanálisis patológico con leucocituria y nitrituria. A la exploración física inicial destaca un supuesto signo del pliegue marcadamente positivo. Ingresa en la unidad neonatal con la sospecha de infección del tracto urinario y durante la evolución presenta proteína C reactiva (PCR) máxima de 32,85mg/l, procalcitonina 2,23 ng/ml, y hemocultivo y urocultivo positivos a Escherichia coli, con evolución favorable bajo tratamiento antibiótico según antibiograma. La ecografía abdominal posnatal confirma la ectasia piélica izquierda (de 11,1mm), con buena diferenciación córtico-medular, con parénquima y cálices normales, sin visualización de uréter proximal. Se realiza cistouretrografía miccional que no muestra reflujo vésico-ureteral.
A pesar de la resolución del cuadro clínico y la normalización de la hidratación es llamativa la persistencia de una piel laxa sin elasticidad aumentada y con pliegues redundantes (fig. 1). Se realiza un estudio de sialotransferrinas y Apo C-III para cribado de defectos congénitos de la glucosilación, que resultan normales. La ecografía transfontanelar confirma los hallazgos prenatales y la resonancia magnética (RM) cerebral detecta también cavum septum pellucidum y cavum vergae, sin alteraciones parenquimatosas evidentes ni otros hallazgos relevantes. La ecocardiografía muestra estenosis ligera de rama pulmonar izquierda. Durante el ingreso presenta un episodio de hipotonía generalizada de pocos segundos con electroencefalograma normal.
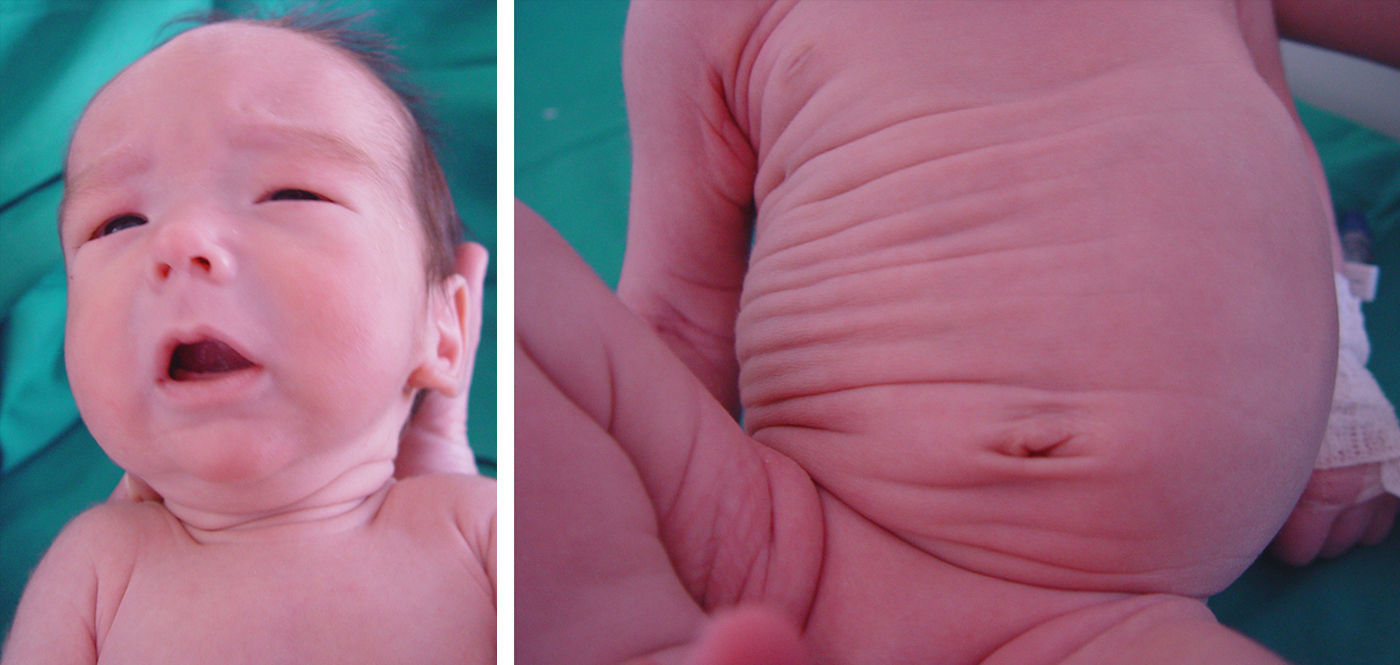
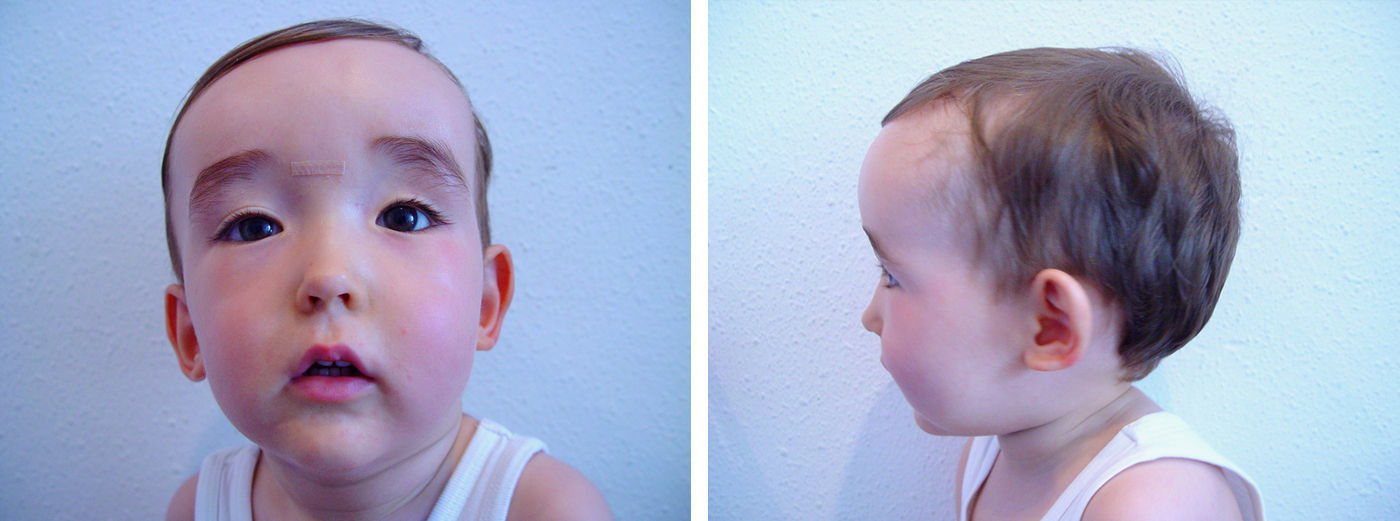
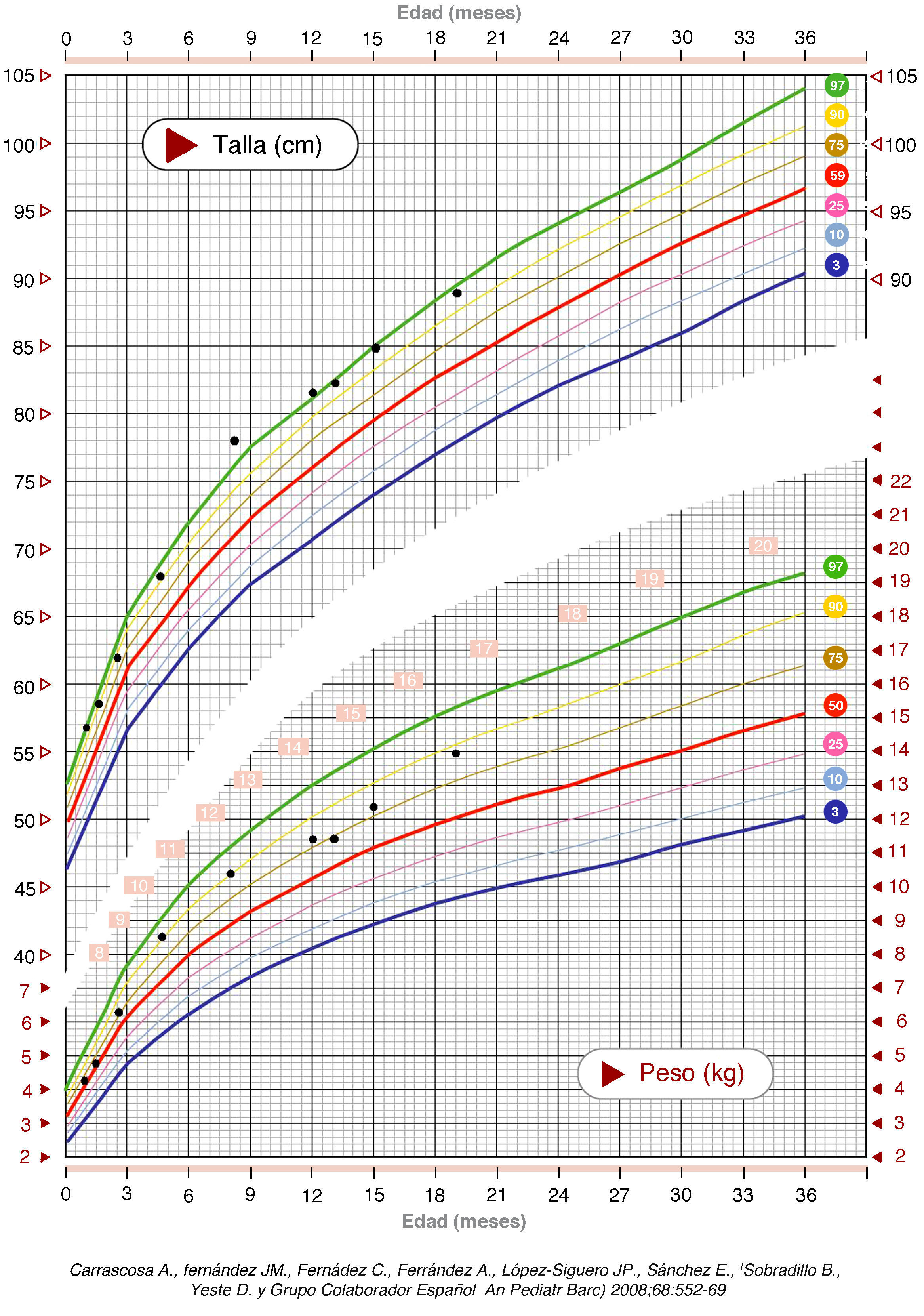
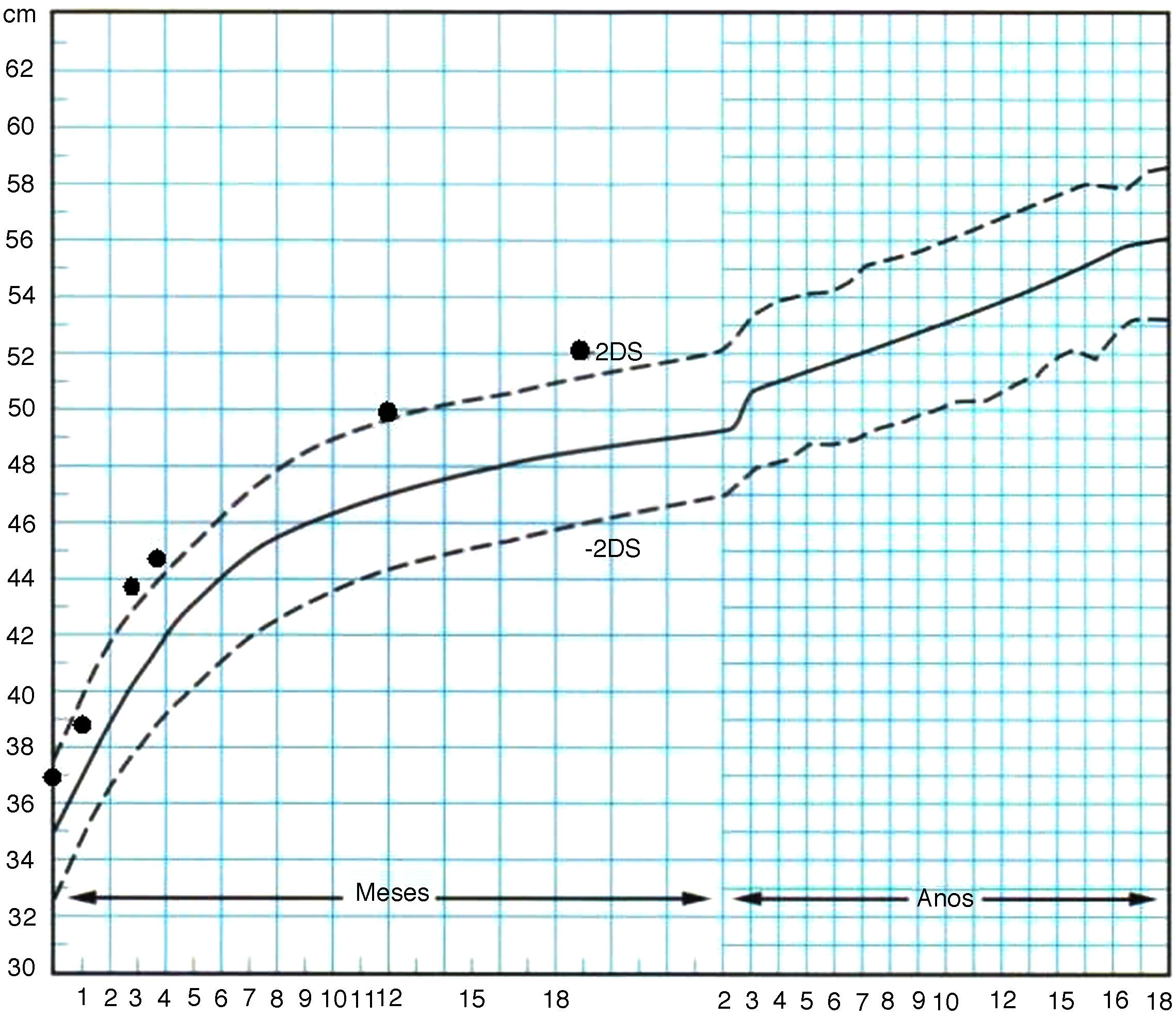
En el seguimiento posterior, persiste inicialmente una cutis laxa llamativa que se normaliza hacia los 3-4 meses, pero persistiendo unos surcos plantares y palmares marcados. Las características fenotípicas resultan cada vez más destacadas con frente prominente asimétrica y pelo escaso con línea de implantación retrasada, ausencia de arcos ciliares, hendiduras palpebrales estrechas, inclinadas hacia abajo y epicantus, raíz nasal aplanada con nariz pequeña, boca pequeña con labio superior fino, paladar ojival, manos y pies grandes, ligera hiperlaxitud articular e hipotonía cervical (fig. 2). Realiza seguimiento para valoración madurativa y acude a centro de estimulación precoz. A los 10 meses presenta una edad ósea de 14 meses. En la somatometría destaca una talla y perímetro cefálico ≥ p97 a partir del mes y 2,5 meses de edad, respectivamente, manteniéndose el peso entre p75-p90 (figs. 3 y 4). Se realiza determinación de IGFBP-3 que es normal (1,74mg/l [0,35 SDS]) y de IGF-1 que es indetectable en dos ocasiones y en el límite bajo de la normalidad a los 20 meses de edad (40 ng/ml [–0,54 SDS]). Se realiza un estudio molecular del gen NSD1, localizado en 5q35, por PCR y secuenciación directa de los 22 exones codificantes (el gen completo contiene 23 exones), detectándose la variante nucleotídica c.4558G>T p.Glu1520Stop en el exón 11. Ésta es una mutación nonsense no descrita previamente, que condiciona la transcripción de una proteína truncada con muy probable carácter patogénico y, por tanto, compatible con el diagnóstico de síndrome de Sotos.
Se define la cutis laxa como una alteración cutánea caracterizada por piel redundante e inelástica, siendo poco frecuentes las formas hereditarias de presentación en el periodo neonatal, con unas 200 familias descritas hasta la fecha5. Dentro del estudio de la cutis laxa congénita con herencia autosómica recesiva o de presentación esporádica se recomienda una investigación amplia para filiar el tipo de alteración, incluyendo el análisis de transferrinas séricas para detectar anomalías de la O- y N-glucosilación de proteínas, y el estudio del patrón de glucosilación de Apo C-III en menores de 6 meses5. En nuestro paciente ambas determinaciones fueron normales, así como la RM cerebral, que en estos pacientes puede mostrar alteraciones en la migración neuronal6,7.
La relación entre síndrome de Sotos y cutis laxa como característica clínica relevante en el periodo neonatal ha sido descrita previamente en 3 pacientes8 diagnosticados por criterios clínicos. Sin embargo, posteriormente a la identificación del gen NSD1, las series de pacientes con alteración genética confirmada no presentan cutis laxa entre sus manifestaciones clínicas4. Tampoco se menciona el síndrome de Sotos entre los que pueden presentar cutis laxa, como son el síndrome de Williams, seudoxantoma elástico, síndromes de Menkes, de Costello o de Kabuki, entre otros5. Sí están descritas otras anomalías que presenta nuestro paciente, como son las de la vía urinaria4,6 o el cavum septum pellucidum y cavum vergae7 como hallazgos en la RM.
A pesar de que hasta la fecha no se han podido identificar marcadores bioquímicos o endocrinológicos específicos del síndrome de Sotos2, es conocida la alteración en la vía de los factores de crecimiento similares a la insulina (IGF) y sus proteínas de transporte (IGFBP)6,9. Se ha postulado el papel de éstas en el crecimiento tanto intraútero como posnatal, sin conseguir resultados ni conclusiones consistentes10. En nuestro paciente se obtuvieron resultados normales de IGFBP-3 y niveles indetectables/bajos de IGF-I en 3 ocasiones.
El gen NSD1 (nuclear receptor SET domain-containing protein-1) fue caracterizado en 2001, y en 2002 se demostró que mutaciones y deleciones de éste eran la causa del síndrome de Sotos, con gran heterogenicidad genética4,9. El gen tiene diversos dominios funcionales, y aunque su papel no está completamente entendido, se creen implicados en la regulación de la cromatina, en la interacción proteína-proteína a nivel del núcleo celular y en la inhibición o activación de la transcripción4,9. El espectro fenotípico de los pacientes con síndrome de Sotos es amplio y algunos estudios han intentado correlacionar las alteraciones genéticas en dicho gen con las manifestaciones clínicas4, sin poder confirmar tal relación, excepto la mayor gravedad de la dificultad de aprendizaje y el menor sobrecrecimiento en los pacientes con microdeleciones 5q35 que incluyen el gen NSD1. Se postula que diferentes factores (estocásticos, ambiente intrauterino, polimorfismos funcionales en genes que interaccionan con el NSD1 y variaciones intrínsecas en la regulación de dianas del gen NSD1) pueden influir en la variabilidad fenotípica del síndrome de Sotos4.
Queremos resaltar la manifestación clínica en forma de cutis laxa presente al nacimiento en nuestro paciente, aunque no podemos afirmar con seguridad si es un hallazgo coincidente o relacionado con la alteración funcional del NSD1.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los padres del paciente por la colaboración abierta en todo momento. Al Dr. J. van Doorn y a su equipo del University Medical Center en Utrecht por su interés y colaboración en la determinación de los valores del eje IGF/IGFBP. A la Dra. Valls por el seguimiento madurativo del paciente y a la Dra. Clofent por el seguimiento integral del paciente en el centro de atención primaria de referencia y a todos los médicos de la Unidad Neonatal (Dr. Coroleu, Dra. Esteban, Dra. Ocaña y Dra. Martínez) y del servicio de pediatría de nuestro hospital.
