
El síndrome de resistencia a las hormonas tiroideas (RHT) es un trastorno hereditario raro causado por mutaciones en el gen del receptor beta de las hormonas tiroideas (THRB)1. Estos cambios en el gen THRB dan lugar a una respuesta alterada a las hormonas tiroideas en tejidos diana y típicamente se presentan con elevación de hormonas tiroideas (triyodotironina [T3] y tiroxina [T4]) asociada a niveles elevados o inapropiadamente normales de tirotropina (TSH)2. El patrón de herencia puede ser autosómico dominante o recesivo, aunque a menudo es dominante3, y su incidencia es de un caso por 40.000 nacidos vivos1. La prevalencia de la RHT debida a cambios en el receptor beta no cambia en base al sexo pero sí varía entre grupos étnicos3,4. El espectro clínico es muy heterogéneo, ya que el síndrome puede ser asintomático o presentarse con síntomas de hipotiroidismo o hipertiroidismo. En casos con antecedentes familiares conocidos es posible el diagnóstico prenatal, lo que facilita la iniciación precoz de la monitorización y el manejo tras el nacimiento.
Presentamos el caso de un neonato con diagnóstico de síndrome de RHT, nacido a las 40semanas de edad gestacional por parto vaginal eutócico en madre de 36años.
La madre tenía antecedentes relevantes de RHT tanto personales como familiares (diagnosticado también en su madre y su hermana). Se encontraba asintomática y no recibía medicación. Se llevó a cabo diagnóstico prenatal mediante biopsia corial durante la primera ecografía prenatal realizada a las 22semanas de gestación, confirmándose que el feto portaba la variante patogénica familiar c.1312C>T (p.Arg438Cys) en el exón10 del gen THRB. El embarazo se desarrolló sin complicaciones.
Tras el nacimiento, los valores Apgar al minuto y a los cinco minutos fueron 9 y 10, respectivamente, y no hubo signos inmediatos de distrés respiratorio o alteraciones hemodinámicas. El examen físico fue anodino, sin rasgos dismórficos o bocio y con un peso al nacer de 3.175g (−0,36SDS).
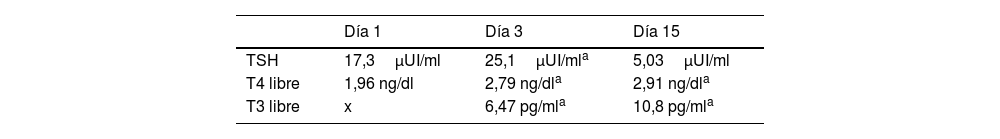
Conociéndose la presencia de la variante patogénica, se envió una muestra de sangre de cordón para realizar pruebas de función tiroidea. Los resultados mostraron un nivel de T4 libre de 1,96ng/dl (rango de referencia, 0,8-2,2)5 con un nivel de TSH de 17,3mUI/l (rango de referencia, 0,5-10,0mUI/l)5. La tabla 1 ilustra la evolución del perfil de función tiroidea en el primer mes de vida.
El neonato recibió el alta a los 2días de vida bajo seguimiento por el equipo de endocrinología pediátrica, con monitorización rigurosa de la clínica, el crecimiento y los valores de las pruebas de función tiroidea. Se mantuvo asintomático, alimentándose bien, con ganancia de peso y crecimiento adecuados. El ecocardiograma y el electrocardiograma a los 27días de vida fueron normales. Dada la estabilidad clínica del paciente y la ausencia de síntomas, no se inició tratamiento inmediato.
El abordaje del síndrome RHT plantea retos singulares, especialmente en recién nacidos y lactantes. Aunque la mayoría de los pacientes con RHT se mantienen asintomáticos, las manifestaciones clínicas pueden ser muy heterogéneas, por lo que se requiere un manejo y una monitorización personalizados. En el presente caso, el diagnóstico prenatal, que es poco frecuente, permitió una monitorización posnatal inmediata, evitándose así posibles retrasos en la detección de disfunción tiroidea y el uso innecesario de medicación.
La principal consideración en el manejo de los neonatos con RHT es la distinción entre la disfunción tiroidea real y el estado fisiológico de RHT, en el que hay elevación de hormonas tiroideas pero la sensibilidad de los tejidos diana se encuentra alterada. La mayoría de los neonatos asintomáticos, como nuestro paciente, no requieren tratamiento. No obstante, es esencial monitorizar estrechamente la función tiroidea, el crecimiento y el neurodesarrollo para garantizar una evolución óptima a largo plazo. Ante cualquier signo de retraso del crecimiento o del neurodesarrollo se debería reevaluar el estado tiroideo y contemplar la necesidad de tratamiento.
Los hallazgos en el caso presentado concordaron con los patrones analíticos característicos de la RHT, en los que coexisten la elevación de la T4 y la T3 libres y la falta de supresión de la TSH. El estudio genético confirmó el diagnóstico prenatalmente, pero la condición clínica del paciente se mantuvo estable. Aunque la penetrancia del síndrome de RHT es alta, su expresión fenotípica es variable, incluso para una misma variante del gen THRB4.
En el caso presentado, la ausencia de síntomas apoya el abordaje conservador actual, en el que la monitorización estrecha es la estrategia principal de manejo durante la infancia.
El manejo a largo plazo de la RHT requiere mantener el eutiroidismo en tejidos en los que la resistencia a las hormonas tiroideas puede ser parcial. En algunos casos podría precisarse administrar dosis suprafisiológicas de hormona tiroidea para suprimir la TSH y asegurar una función tiroidea adecuada en tejidos con sensibilidad reducida.
En conclusión, el caso presentado subraya la importancia de los antecedentes familiares en el diagnóstico precoz de trastornos genéticos como el síndrome de RHT. Este caso es inusual, dado su diagnóstico prenatal, y es uno de los pocos reportados con manejo de la enfermedad en los primeros meses de vida. El abordaje de los pacientes con RHT, especialmente los diagnosticados a edades muy tempranas, presenta retos considerables. Distinguir entre una disfunción tiroidea real y el estado fisiológico de resistencia es crucial y requiere una monitorización continua e individualizada. Aunque muchos neonatos asintomáticos, como nuestro paciente, no requieren tratamiento inmediato, es esencial mantener un seguimiento riguroso del crecimiento y de la función tiroidea para garantizar una evolución favorable a largo plazo. Por lo tanto, el abordaje de estos pacientes no puede subestimarse, ya que la complejidad y la heterogeneidad clínica del síndrome precisan un manejo cuidadoso a cargo de un equipo de endocrinología pediátrica y seguimiento de por vida.

