



Presentamos el caso de una paciente joven con diagnóstico clínico y genético de síndrome de Kearns-Sayre (SKS). Como hallazgos, destacaban blefaroptosis, limitación de la motilidad extrínseca ocular, retinopatía en sal y pimienta así como insuficiencia mitral leve. La anatomía patológica indicaba citopatía mitocondrial y el estudio genético detectó deleción y duplicación en heteroplasmia del ADN mitocondrial.
El SKS es un raro trastorno neuromuscular, caracterizado por la tríada oftalmoplejía externa progresiva, retinopatía pigmentaria y bloqueo cardíaco. La detección temprana del SKS es clave para evitar potenciales complicaciones cardíacas.
The clinical case and genetic diagnosis of Kearns-Sayre syndrome (KSS) is described in a young patient. The findings included: ptosis, ocular motility disturbances, pigmentary retinopathy, as well as mitral insufficiency. A muscle biopsy revealed mitochondrial cytopathyand heteroplasmic mitochondrial DNA deletions.
KSS is a rare neuromuscular disorder defined by a characteristic triad of progressive external ophthalmoplegia, pigmentary retinopathy and atrioventricular block. Early detection is essential to avoid potential cardiac complications.
El síndrome de Kearns-Sayre (SKS) fue descrito por primera vez en 1958. Es un trastorno neuromuscular causado por defectos genéticos en el ADN mitocondrial (mtADN). Los pacientes afectados presentan: oftalmoplejía externa progresiva crónica, retinopatía pigmentaria, bloqueo cardiaco y ataxia cerebelosa1. Los síntomas de la enfermedad aparecen generalmente en la adolescencia y raramente en la edad adulta. Presentamos el caso de una paciente que presenta SKS con una evaluación oftalmológica completa, así como el análisis de su mtADN.
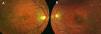
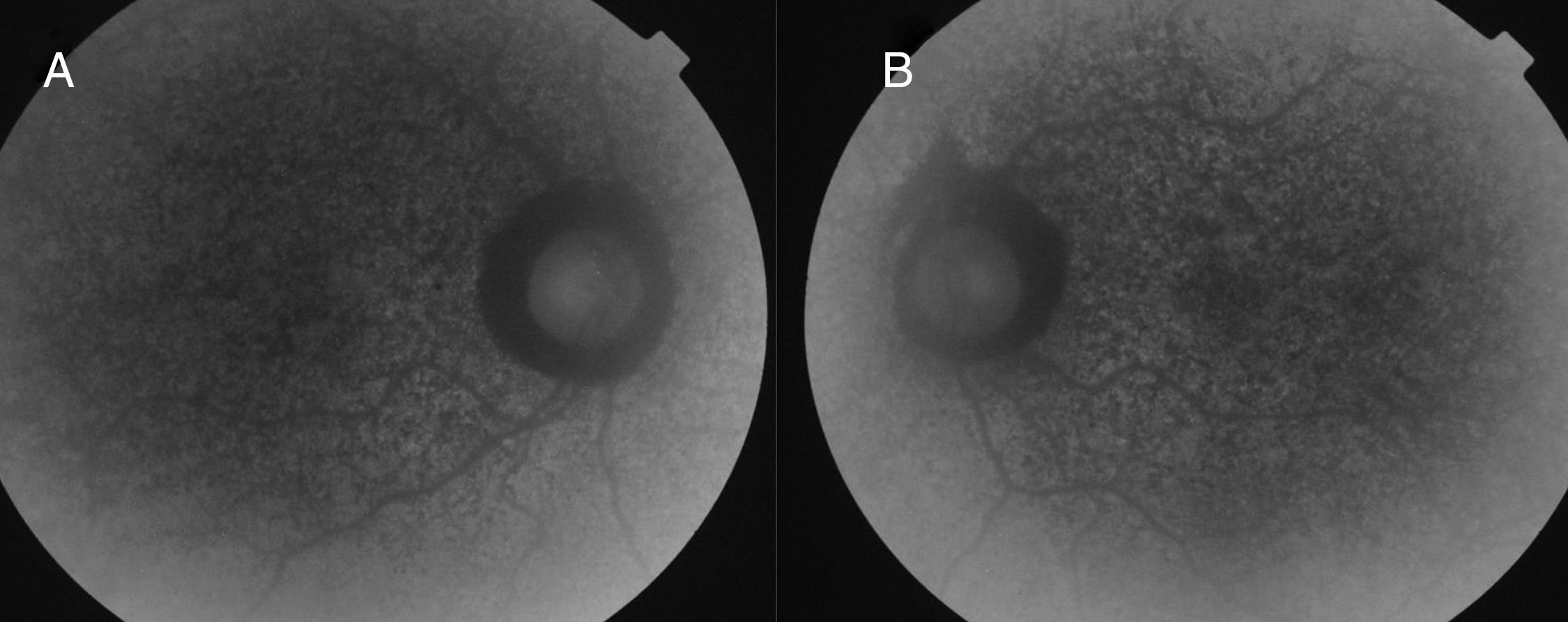
Caso clínicoPaciente de 15 años, remitida a nuestro hospital por presentar ptosis y limitación de los movimientos oculares. La paciente presentaba una buena visión en ambos ojos (20/20). La exploración de la motilidad ocular mostró marcada limitación de la motilidad ocular extrínseca con exotropia de 15° del ojo izquierdo (OI). Presentaba una blefaroptosis de grado moderado-grave con oclusión parcial de la pupila, mayor en ojo derecho (OD). La función del elevador era débil, pero conservada en ambos ojos. La exploración del segmento anterior era normal. En el examen del fondo de ojo, se pudieron apreciar cambios pigmentarios con un patrón en sal y pimienta (fig. 1A y B). La autofluorescencia evidenció una atrofia del epitelio pigmentario con zonas de hiper e hipoautofluorescencia (fig. 2A y B). La electrorretinografía demostró una disminución de las ondas a y b a respuesta de estímulos luminosos, previa dilatación y adaptación a la luz. En el estudio con potenciales visuales evocados se observó un aumento en el tiempo de conducción. Se observó un aumento en la concentración de transaminasa glutamicoxalacética, transaminasa glutámico pirúvica, lactatodeshidrogenasa y creatincinasa en la bioquímica sanguínea. La paciente presentaba, además, una insuficiencia mitral leve con prolapso. La anatomía patológica de una biopsia de tejido muscular a nivel de cuádriceps indicó una citopatía mitocondrial con presencia de fibras de tipo 1, con ausencia de la actividad enzimática y un aumento del tamaño mitocondrial. Se realizó un diagnóstico genético de confirmación mediante la detección, a partir de una muestra sanguínea, de una deleción en el mtADN de esta paciente y duplicación en heteroplasmia.
Discusión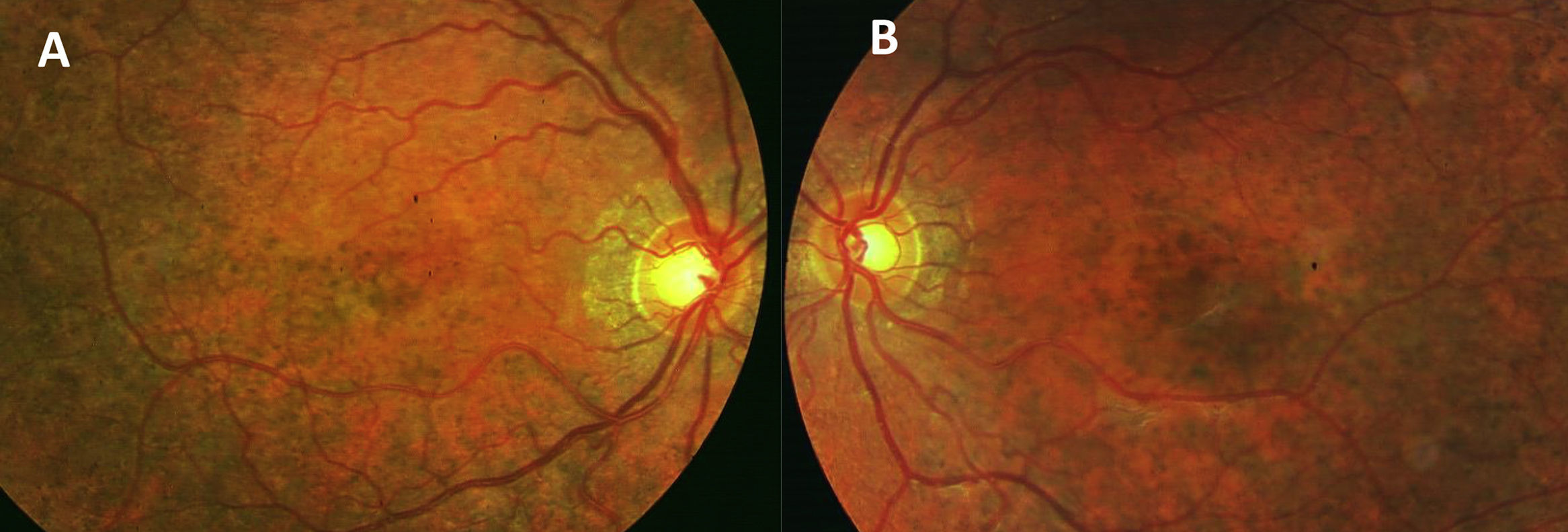
El SKS es una rara enfermedad multisistémica que afecta preferentemente a pacientes menores de 20 años. Es una citopatía mitocondrial en relación con deleciones del mtADN de los músculos estriados, del miocardio, del sistema nervioso central o periférico, de la piel y del epitelio pigmentario de la retina (EPR). En la biopsia muscular es típica la presencia de las fibras «ragged red», que corresponden a fibras musculares con acumulación de mitocondrias anormales2.
La ptosis y la oftalmoplejía son los signos oftalmológicos más comunes. La disminución de la agudeza visual es variable en función del grado de deterioro en la retina. El examen histopatológico de esta enfermedad indica que el EPR es el que se afecta primero, seguido por los fotorreceptores de la retina y la coriocapilar3. La atrofia del EPR predomina más en la periferia de la retina que en el polo posterior. En esta paciente, encontramos heteroplasmia en el análisis genético, una mezcla de mtADN mutante con silvestre en la misma célula. Esta relación de ADN mutado y ADN silvestre es de gran importancia, ya que nos va a determinar la severidad del fenotipo. La buena agudeza visual de la paciente, inusual en estos casos, puede ser explicada por la existencia de esta heteroplasmia4.
Actualmente, no existe ningún tratamiento específico eficaz para el SKS, el tratamiento es paliativo y de apoyo para las condiciones clínicas asociadas. Algunos pacientes con miopatía se benefician con el uso de la coenzima Q10, especialmente, aquellos con mutaciones que producen la reducción de la síntesis de esta proteína5.
El pronóstico y la supervivencia de esta enfermedad están ligados en muchas ocasiones a la evolución de las alteraciones cardíacas6. En lo que se refiere a las manifestaciones oftalmológicas, la pérdida de visión debida a la degeneración pigmentaria de la retina es menos grave que la asociada a la retinosis pigmentaria típica, siendo muy raro que presente ceguera nocturna o constricción del campo visual3. A diferencia de la mayoría de los casos de SKS, que cursan con disminución de la agudeza visual, en esta paciente se encuentra conservada. Esta es una forma de presentación muy poco frecuente, según lo revisado en la literatura7, y refleja la variabilidad de manifestaciones del SKS en relación con el tipo de mutación mitocondrial y su grado de expresividad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
