




El síndrome de Griscelli es una enfermedad autosómica recesiva muy rara, que fue descrita por primera vez en 19781. Se han comunicado desde entonces menos de un centenar de casos, la mayoría en lactantes y escolares. Se presenta el siguiente caso clínico, esperando enriquecer el conocimiento de este síndrome.
Una paciente nativa boliviana de 15 meses de edad es admitida en el servicio de infectología debido a fiebre y distensión abdominal de 1 mes de evolución. Era producto de un cuarto embarazo de un matrimonio sin consanguinidad, sin antecedentes patológicos prenatales, perinatales y familiares de importancia.
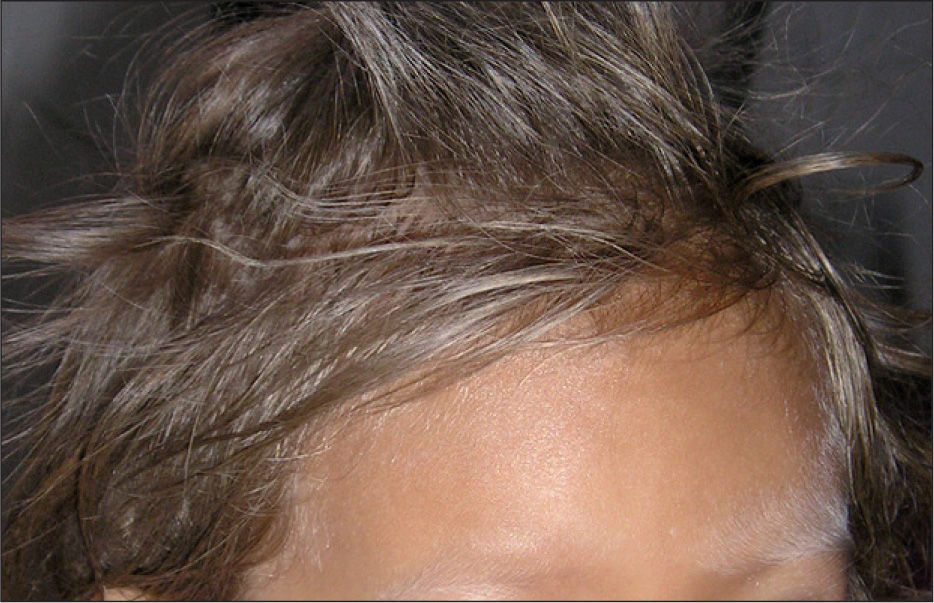
En el examen físico la paciente presenta un peso de 8,5kg y una talla de 75cm que corresponden a una desnutrición de primer grado según las tablas antropométricas. Llama la atención la presencia de cabello, pestañas y cejas de color plateado (fig. 1). Otras manifestaciones clínicas incluyen una linfadenopatía generalizada, hepatoesplenomegalia, petequias en todo el cuerpo y un desarrollo neurológico normal para la edad.
Se consideró un síndrome de los pelos plateados y un síndrome hemofagocítico clínicamente, pero fue necesario descartar primero algunas patologías que se acompañen con hepatoesplenomegalia y adenopatías generalizadas con fiebre.
El hemograma muestra una hemoglobina de 8,4g/dl, leucocitos totales de 6.200/μl, con polimorfonucleares del 36 %, linfocitos del 60 %, eosinófilos del 3 % y basófilos del 1 %. No se encontró vacuolización citoplasmática en los leucocitos en el frotis de sangre periférica. El recuento plaquetario muestra una plaquetopenia de 45.000/μl. Las pruebas de función hepática fueron normales. Los tiempos de coagulación se encontraban dentro de rangos normales, pero el fibrinógeno estaba descendido 48mg/dl. La electroforesis de proteínas no mostró alteraciones. Las radiografías de tórax, huesos largos y la ultrasonografía abdominal no mostraron datos patológicos más que la hepatoesplenomegalia.
Se realizó de manera adicional una aspiración, cultivo y biopsia de médula ósea sin encontrarse datos patológicos. La tinción de mieloperoxidasa de células de médula ósea resultó negativa. El perfil lipídico mostró hipertrigliceridemia (256mg/dl) e hipocolesterolemia (58mg/dl).
Se descartaron la infección por citomegalovirus, virus de la inmunodeficiencia humana, virus de Epstein Barr, toxoplasmosis, malaria, tripanosomiasis americana, leshmaniasis visceral y tuberculosis.
El examen con microscopia de luz óptica de un fragmento de pelo mostró cúmulos pigmentarios, de forma y tamaños irregulares, distribuidos de manera uniforme (fig. 2). Este dato nos abrió la sospecha de los síndromes de Griscelli, Chediak-Higashi y Elejalde.

El síndrome de Chediak-Higashi se descartó por la ausencia de gránulos intracitoplasmáticos celulares en el frotis de sangre periférica y mielograma. La ausencia de alteración neurológica excluyó un síndrome de Elejalde, lo que sugería como diagnóstico por exclusión un síndrome de Griscelli.
Durante su hospitalización la paciente adquirió una infección pulmonar que fue tratada con antibióticos sistémicos con buena respuesta. Se consideró la utilización de corticoides, quimioterapia y trasplante de médula ósea como opciones terapéuticas en el síndrome de Griscelli, pero no se consiguió el consentimiento de la familia debido a limitantes económicos.
La hemoglobina, plaquetas y leucocitos fueron disminuyendo en controles posteriores, a pesar de las numerosas transfusiones de sangre y plaquetas que recibía la paciente. El tiempo de coagulación fue descendiendo a pesar de la administración de plasma fresco congelado y vitamina K, llevando a una hemorragia grave incontrolable y que acabó con la vida de la paciente después de 21 días de hospitalización.
No se realizó examen cromosómico, pero sí autopsia de hígado y bazo, los cuales mostraban datos compatibles con hemofagocitosis encontrados en el síndrome de Griscelli tipo 2 y Chediak-Higashi.
El diagnóstico diferencial del síndrome de los pelos plateados incluye tres entidades clínicas: el síndrome de Elejalde, el cual se manifiesta predominantemente con alteraciones neurológicas; el síndrome de Chediak-Higashi con inmunodeficiencia grave y presencia de gránulos intracitoplasmáticos leucocitarios y por último el síndrome de Griscelli que varía clínicamente según la mutación genética encontrada2.
El síndrome de Griscelli de tipo 1 se presenta con hipomelanosis y déficit neurológico primario; el tipo 23, causado por mutación en el gen RAB27A, se manifiesta con hipomelanosis, deterioro inmunológico y manifestaciones de un síndrome hemofagocítico y el tipo 3 muestra simplemente manifestaciones cutáneas y se debería a mutaciones en los genes MYO5A4.
Algunos autores no distinguen diferencias clínicas entre el síndrome de Griscelli de tipo 1 y Elejalde, e incluso se ha postulado que sean una misma enfermedad5,6. La evidencia molecular sustenta la posibilidad de tratarse de dos patologías distintas cromosómicamente relacionadas7.
Este caso en particular muestra muchos criterios clínicos de un síndrome de Griscelli de tipo 2 el cual muestra muchos datos clínicos e inmunohematológicos con un alto índice de mortalidad, como fue el caso de esta paciente.
