
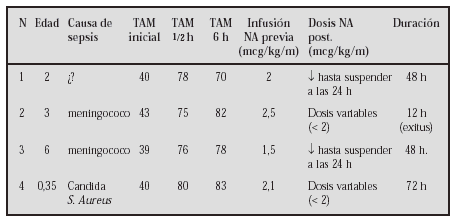
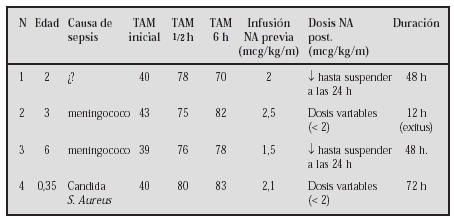
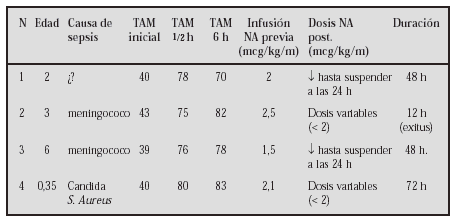
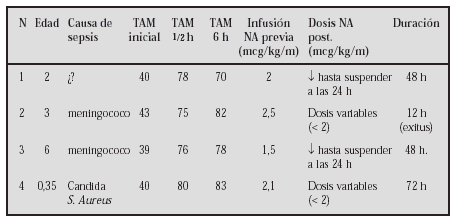
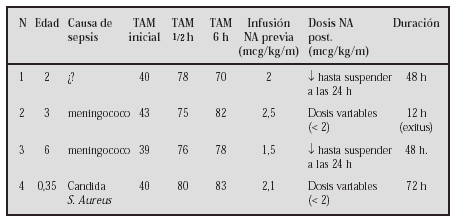
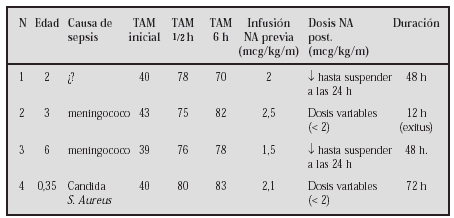
GASTROENTEROLOGÍA Y HEPATOLOGÍA
P172 DIAGNÓSTICO EN LA ATRESIA DE VÍAS BILIARES. NUESTRA EXPERIENCIA. 08:30 h
César Emilio Silverio García, Wladimiro García Pérez
Hospital Pediátrico Docente "William Soler", La Habana (Cuba).
El diagnóstico precoz del lactante con atresia de vías biliares es de vital importancia para el éxito de la intervención quirúrgica por lo que nuestro estudio esta encaminado a realizar con la mayor prontitud y seguridad el diagnóstico en lactantes colestásicos que presentan esta patología. Para esto efectuamos una investigación retrospectiva y prospectiva que abarca desde 1990 hasta el 2003 en la que valoramos en cada paciente los datos generales, clínica, analítica, ecografía abdominal y resultado del estudio laparoscópico sin o con colangiografía. En los datos generales analizamos sexo, raza, momento del nacimiento; en la clínica, el antecendente del peso al nacer, aparición de la sintomatología colestasica, hallazgos al examen físico; en la analítica, comportamiento de la hemoglobina, coagulación y GTP; en la ecografía, la presencia o ausencia de vesícula biliar y en el estudio laparoscopico, el aspecto macroscopico del hígado y confirmación colangiografica de la enfermedad. En nuestro grupo se observó la atresia biliar con mayor frecuencia en los lactantes del sexo femenino, raza blanca, nacidos en verano y con buen peso al nacer. Por lo general el íctero apareció después de los 14 días de nacido y el hallazgo más llamativo al examen físico además del íctero es la presencia de hepatomegalia dura. La mayoría de nuestros pacientes presentaban anemia, la elevación de la GTP fue moderada y no existía coagulopatia. La presencia de vesícula biliar no descarta la enfermedad. El estudio laparoscopico nos permitio conocer el estado de afectación del hígado: sin cirrosis, en evolución a la cirrosis, y con cirrosis, además, valorar la presencia o no de signos de hipertensión portal. Se demuestra la utilidad de la colangiografia translaparoscopica como método diagnostico confirmatorio de esta enfermedad.
P173 MACROAMILASEMIA COMO FACTOR DE CONFUSIÓN EN UN PACIENTE CON DOLOR ABDOMINAL AGUDO. 08:35 h
Cristina Olivas López de Soria, Javier Álvarez-Coca González, Mercedes Gómez Manchón, Álvaro Lassaletta Atienza, M. Pilar González Santiago, Gloria López Lois, María Penín Antón, Jimena del Castillo Peral, Inés Merino, José Enrique García de Frías
Hospital Príncipe de Asturias, Alcalá de Henares (Madrid).
Introducción: La macroamilasemia (MA) es una condición en la que la amilasa sérica se une a ciertas proteínas formando un complejo de alto peso molecular, lo que supone un obstáculo para su excreción renal. Su diagnóstico de sospecha se basa en la presencia de hiperamilasemia persistente con amilasuria disminuida, en ausencia de enfermedad renal y sin causas que la justifiquen. La confirmación diagnóstica se realiza con la electroforesis y la cromatografía en columna. La etiología es desconocida y la prevalencia oscila entre un 1,6% y 4,5% en adultos hiperamilasémicos, siendo excepcional en niños. Se ha observado asociada a tumores, enfermedad hepatobiliar, enfermedades autoinmunes y, últimamente, a la enfermedad celiaca. Es asintomática, aunque puede ser causa de confusión diagnóstica con la pancreatitis en pacientes con dolor abdominal agudo.
Caso clínico: Varón de 9 años con dolor abdominal cólico, fiebre elevada y vómitos biliosos de 48 horas de evolución. Deposiciones pastosas verdosas. No antecedente traumático ni disuria. Antecedentes personales: dolor abdominal periumbilical y nauseas de cinco meses de evolución. Exploración física: abdomen discretamente doloroso a nivel epigástrico y periumbilical. Analítica: elevación progresiva de la amilasa sérica (1.162) y lipasa elevada (185). Ecografía abdominal normal. Coprocultivo positivo para Campylobacter jejuni. Se diagnosticó de pancreatitis aguda leve asociada a infección entérica por C. Jejuni. En el seguimiento posterior durante un año, han persistido cifras de amilasemia entre 775 y 910 U/l, con amilasuria baja, permaneciendo asintomático. Se determinaron las isoenzimas de amilasa, hallándose un porcentaje normal de la fracción P2 y un porcentaje elevado de la fracción S2, compatible con macroamilasa.
Conclusiones: La MA debe tenerse en cuenta en el diagnóstico diferencial de hiperamilasemia persistente sin signos clínicos de pancreatitis o inflamación de las glándulas salivales. La MA y la pancreatitis pueden coexistir, por lo que la determinación de isoenzimas de la amilasa y la lipasa pueden ayudar a excluir una pancreatitis. En todo paciente con dolor abdominal agudo e hiperamilasemia, la observación de una amilasuria disminuida nos debe hacer pensar en la presencia de una MA.
P174 COLECISTITIS ACALCULOSA POR SALMONELLA GRUPO C. 08:40 h
Luis Padilla Hernández, Juan Luis Santos Pérez, Miguel Ángel López Casado, María Rodrigo Moreno, Alicia Quesada Alguacil, Emilia Urrutia Maldonado, Purificación Cárdenas Guerrero, M. Rosario Benavides Román, Luis Carlos Ortiz González, Luis Ortega Martos
Hospital Virgen de las Nieves, Granada.
La colecistitis es una entidad menos frecuente en la edad pediátrica que en el adulto. Por cada 1.000 casos en adultos hay 1,3 casos pediátricos. La colecistitis acalculosa (CA) ocurre en un 2-17% de los casos de colecistitis en adultos, mientras que entre un 30-50% de los casos infantiles se producen en ausencia de cálculos, siendo secundarias a cuadros infecciosos en la mayoría de los casos. Presentamos un caso de un paciente ingresado en la UCI pediátrica de nuestro centro por una CA secundaria a infección por salmonella grupo C. Varón de 6 años, previamente sano, que presenta fiebre elevada, dolor abdominal, vómitos y diarrea escasa de 72 horas de evolución. En las últimas horas empeora su estado general y aumenta el dolor abdominal, apareciendo ictericia y coluria. En las exploraciones complementarias realizadas destaca la presencia de derrame pleural bilateral en la Rx de tórax y en la ecografía abdominal engrosamiento marcado de la pared vesicular, sin dilatación de vías biliares ni presencia de cálculos. Ante la instauración progresiva de estado de shock, se traslada a la UCIP de referencia. A su ingreso presenta taquicardia e hipotensión, siendo precisa corrección de volemia y administración de drogas vasoactivas. Presenta síntomas compatibles con SDRA por lo que se procede a intubación y ventilación mecánica. Desarrolla posteriormente disfunción hepática, alteración de la coagulación e insuficiencia renal. En el hemocultivo y coprocultivo se aísla Salmonella grupo C. Persiste imagen de engrosamiento de la pared vesicular (máximo 6 cm en controles). Tras la instauración de antibioterapia con amoxicilina-clavulánico, se produce la mejoría clínica del paciente. Se suspende la ventilación mecánica a los 10 días y se da de alta a los 17 días. La ecografía de control muestra vesícula de tamaño y ecogenicidad normales, sin dilatación de vías biliares. La CA es una enfermedad grave que se caracteriza por la presencia de fiebre, ictericia y dolor abdominal. La ecografía es fundamental para su reconocimiento, mostrando engrosamiento de la pared vesicular en ausencia de litiasis biliar. El retraso en el diagnóstico y en el tratamiento es causa de elevada mortalidad, recomendando muchos autores el tratamiento quirúrgico precoz. No obstante, otros trabajos muestran evolución favorable con tratamiento conservador, en aquellos casos en que la situación del paciente sea crítica.
P175 ENFERMEDAD INFLAMATORIA INTESTINAL EN LA EDAD PEDIÁTRICA EN EL ÁREA SANITARIA DE FERROL. 08:45 h
Ana Isabel García Villar, Carmen Gómez Lado, Clara Fariña Candal, Gloria Landín Iglesias, Marta Carballal Mariño, Maravillas Santos Tapias, Ramón Fernández Prieto
Hospital Arquitecto Marcide-Profesor Novoa Santos, Ferrol (A Coruña).
Objetivo: Análisis de aspectos epidemiológicos, clínicos, antropométricos, nutricionales, analíticos e histológicos.
Material y métodos: Estudio retrospectivo mediante revisión de historias clínicas de pacientes diagnosticados de enfermedad inflamatoria intestinal (EII) desde 09-1994 a 09-2003, recogiéndose características epidemiológicas (edad, sexo, origen poblacional, antecedentes familiares), clínicas (presentación, tiempo de evolución), antropométricas, bioquímicas, radiológicas, endoscópicas, histológicas y hallazgos en densitometría ósea.
Resultados: Se incluyeron 13 pacientes (10 varones y 3 mujeres), edad media al diagnóstico de 8 años (rango 3,5-13 años). El 85% proceden de medio urbano. Presentaba antecedentes familiares de EII uno de los pacientes. El 54% presentan enfermedad de Crohn (EC), 15% colitis ulcerosa (CU) y 31% colitis indeterminada (CI). Forma de presentación fue diarrea sanguinolenta y dolor abdominal en 100% de los pacientes con CU. 100% de los casos de EC presentaron diarrea (71% no sanguinolenta), los pacientes con CI el 50% presentaban retraso póndero-estatural. Tiempo de evolución al diagnóstico fue 17 días en CU, 3 meses en EC y 14 meses en CI. Al diagnóstico 77% presentaban peso menor del percentil 50 (20% menor del percentil 3), 53,8% talla menor del percentil 50 (14% menor percentil 3), 92% índice de masa corporal menor percentil 50 (16,5% menor percentil 3). Estudio endoscópico e histológico se realizó en 100% casos. 86% de los pacientes con EC presentaban localización ileocólica y 14% ileal. La evolución de la velocidad de crecimiento fue favorable en 84,6% pacientes y del índice de masa corporal en 70%. Presentaron manifestaciones extraintestinales 31% y complicaciones 69%.
Conclusiones:1) Tendencia a presentarse la EII en edades más tempranas. 2) Más frecuente en población urbana. 3) La forma de presentación clínica más frecuente en la EC el la tríada clásica de dolor abdominal, diarrea y pérdida peso, siendo en la CU la diarrea sanguinolenta. 4) Localización más frecuente de la EC es ileocólica. 5) La osteoporosis es una complicación frecuente. 6) Importante el diagnóstico precoz para evitar repercusión en crecimiento. 7) En estudio de talla baja, considerar EII como posible causa.
P176 DEFICIENCIA DE ZINC SINTOMÁTICA EN PREMATUROS ALIMENTADOS CON LACTANCIA MATERNA. 08:50 h
Gemma Arca Díaz, Jesús Carnicer de la Pardiña, Eulàlia Baselga Torres, Israel Anquela Sanz, Raúl Morales Prieto, Isabel Badell Serra, José Cubells Rieró
Hospital de la Santa Creu y Sant Pau, Barcelona.
Introducción: La deficiencia de zinc (Zn) en prematuros alimentados con lactancia materna es un trastorno raro, poco frecuente en la literatura, causado por unos niveles bajos de este oligoelemento en leche materna. Los prematuros son más vulnerables a desarrollar un déficit de zinc al producirse la máxima retención de este oligoelemento en los últimos meses de gestación y su intestino inmaduro es incapaz de reabsorberlo. La presentación clínica es similar a la acrodermatitis hereditaria enteropática (AE). El tratamiento de la forma adquirida se basa en la administración de suplementos orales de Zn.
Caso clínico: Lactante varón de cuatro meses de edad, con antecedente de prematuridad de 27 semanas de gestación que fue alimentado durante los primeros ocho días con nutrición parenteral y posteriormente con lactancia materna exclusiva. Inició a los dos meses un cuadro de diarrea persistente, vómitos y lesiones eczematosas de la piel, acra y periorificial.
Resultados: La analítica mostró bajos niveles de zinc en el suero del lactante (3 µmol/l) y en la leche materna (8 µmol/l), con un nivel normal de zinc en el suero materno (17 µmol/l). Se trató con suplementos de sulfato de zinc vía oral (2 mg/Kg), remitiendo en un mes la clínica. En el control evolutivo de más de un año no han recidivado las lesiones tras supresión del tratamiento.
Conclusión: El déficit de Zinc en la lactancia materna da lugar a una deficiencia de este oligoelemento de forma adquirida que suele ocurrir en prematuros alimentados con lactancia materna y también se asocia la nutrición parenteral. Algunos autores proponen la determinación de Zn en pacientes pretérmino alimentados con lactancia materna exclusivamente a los 3 ó 4 meses de vida. El déficit de Zn en la leche materna podría ser producido por un defecto en la transferencia de zinc del suero materno a la leche o debido a la secreción defectuosa mamaria.
P177 ENFERMEDAD CELÍACA ASOCIADA A ENFERMEDADES AUTOINMUNES. 08:55 h
Elena Lucas Sáez, Beatriz Tresaco Benedi, Aurora Lázaro Almarza, José Luis Olivares López, Josefa López Moreno
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción: La Enfermedad Celíaca (E.C.) es una enteropatía sensible al glúten, caracterizada por un amplio espectro de manifestaciones clínicas. Una alta proporción de enfermos celíacos asocian enfermedades autoinmunes (E.A.). El riesgo de aparición de anticuerpos contra glándulas endocrinas aumenta con el diagnóstico tardío y el mal cumplimiento de la dieta libre de glúten.
Objetivo: Las E.A. pueden ser formas de presentación no clásica de la E.C. Por ello debemos pensar que bajo una E.A. puede subyacer una E.C. no diagnosticada, y ante una E.C. con manifestaciones autoinmunes puede existir un mal seguimiento de la dieta sin glúten.
Material y métodos: Presentamos 8 casos de E.C. con diagnóstico clínico, serológico y anatomoclínico según criterios de la ESPGAN en 1989, que asociaron una E.A.
Resultados: Se muestran en una tabla donde se detallan las edades y formas de presentación de una y otra enfermedad, los antecedentes familiares y los HLA. Uno de los pacientes tenía también Síndrome de Down.
Conclusiones: Las E.A. que con mayor frecuencia se asociadan a E.C. son la Diabetes Mellitus I y la tiroiditis autoinmune. Menos habituales son la artritis seronegativa y la nefropatía IgA. También es frecuente que el Síndrome de Down asocie E.C., aunque el diagnóstico puede retrasarse por ser atribuidos ciertos síntomas intestinales a la trisomía. El glúten podría representar un factor de comienzo o mantenimiento de estos procesos autoinmunes. Además de aumentar la prevalencia, el no cumplimiento de la dieta empeora el pronóstico. En el caso de la diabetes, el debut es más temprano; y en el de la artritis, ésta es clínicamente más severa. En la mitad de nuestros casos, las E.A. debutaron en la adolescencia, edad en la que son comunes las transgresiones dietéticas. Mientras que la dieta sin glúten ha negativizado los marcadores antiendomisio y antitransglutaminasa en nuestros pacientes, el resto de anticuerpos (anti-islote pancreático y antitiroideos) no lo han hecho. De ahí la importancia de instaurar de forma temprana una dieta sin glúten, recién hecho el diagnóstico, y así disminuir la asociación con otras enfermedades.
P178 REFLUJO GASTROESOFÁGICO, ACROPAQUIAS, ANEMIA E HIPOPROTEINEMIA: TRÍADA DE HERBST. 09:00 h
Julio Guerrero Vázquez, Julio Guerrero Fernández, Marta Taida García-Ascaso, José Luis Luengo Casasola, Carlos Collantes García, Mariano Vicente Cuevas
Hospital del S.A.S. Punta de Europa, Algeciras (Cádiz) y Hospital Materno Infantil La Paz, Madrid.
Introducción: Las acropaquias constituyen un hallazgo clínico llamativo que habitualmente relacionamos con neumopatías y/o cardiopatías causantes de hipoxemia. También de han descrito asociadas a patología digestiva (hepatopatías, E. de Crohn, etc.) siendo excepcionales las de origen esofágico (generalmente neoplasias). Son múltiples las situaciones patológicas derivadas de la existencia del Reflujo gastroesofágico (RGE) pero muy infrecuente y escasamente conocida la denominada triada de Herbst (TH) que junto a las acropaquias incluye anemia e hipoproteinemia que, característicamente, desaparecen cuando se corrige el RGE.
Caso clínico: Niña de 6 años sin antecedentes familiares, gestacionales o perinatales de interés. Desde las primeras semanas de vida, vómitos alimenticio-mucosos y procesos catarrales recurrentes. Al año de edad evidente hipocratismo digital. Los vómitos fueron atenuándose, persistiendo episodios de tos acompañados de la emisión de secreciones mucosas surcadas, ocasionalmente, por estrías sanguinolentas. A los síntomas citados se añaden en el último mes dolor epigástrico y retroesternal. Examen físico: Obesa, discreta palidez, dedos en palillo de tambor, ausencia de alteraciones osteoarticulares, auscultación cardiopulmonar normal. Exámenes complementarios: Hemoglobina 8,2 g/dl, hipocromía y microcitosis; series blanca y plaquetaria normales; sideremia 15 mcg/dl, ferritina 7 ng/l; normales; proteínas totales 5,1 g/dl, albúmina 55%; hemorragias ocultas en heces positivo; ionotest repetidamente negativo; parámetros de función pancreática y respiratoria normales; RX y TAC torácicos normales; RX mano, articulaciones mayores y huesos largos, sin artritis ni periostitis; esofagograma: severo RGE asociado a hernia hiatal; esofagoscopia: signos de intensa esofagitis confirmada histológicamente. Los padres rechazaron la realización de pH-metría esofágica y las investigaciones radioisotópicas para detectar enteropatía pierde proteínas. Ineficacia de las medidas conservadoras (gastroquinéticos, bloqueantes H2, ferroterapia). Tras la corrección quirúrgica (fundoplicatura) rápida mejoría clínica y normalización analítica. Cinco meses más tarde las acropaquias habían desaparecido.
Comentarios: La TH es una excepcional manifestación de RGE. Se analiza la etiología de las acropaquias y su patogenia. No existe explicación convincente para las desarrolladas en relación con RGE no complicado con neumopatía/hipoxemia.
P179 ACALASIA EN LA INFANCIA: A PROPÓSITO DE UN CASO. 09:05 h
José María Lloreda García, Pascual Caballero Fernández, Carlos Sierra Salinas, Antonio Jurado Ortiz
Hospital Materno Infantil Carlos Haya, Málaga.
La acalasia es una enfermedad neuromuscular del esófago caracterizada por falta de relajación del esfínter esofágico inferior tras la deglución, tono elevado del mismo y debilidad o ausencia de ondas peristálticas esofágicas. Es una enfermedad rara en niños (2-3% de los casos). Presentamos el caso de una niña de 8 años, cuarto caso en nuestro hospital en 20 años.
Caso clínico: Paciente de 8 años remitida por la unidad de Salud Mental por un cuadro de 4 meses de evolución caracterizado por vómitos durante las comidas o inmediatamente después, de contenido alimentario, pérdida de peso de 6 kg y alteración del carácter (menos interesada por el juego, más callada). Un año antes, ingreso por dificultad respiratoria, evidenciándose atelectasias en lóbulo superior y medio derechos, secundarios a tapones mucosos y gran inflamación mucosa. En la exploo0ración clínica destaca peso y talla en percentil 25. Durante su estancia en planta se provocó el vómito en varias ocasiones. Se le practicó un TC craneal que fue normal y un tránsito esofagogastricoduodenal en el que se observa un esófago dilatado sin relajación del cardias y pobre paso de contraste al estómago. Se realiza una endoscopia para descartar otros procesos y se evidencia un esófago dilatado en toda su longitud, con restos alimentarios, cardias cerrado, pero que permite el paso del endoscopio y esofagitis y gastritis mínimas. Se practica una manometría esofágica compatible con acalasia, corrigiéndose posteriormente con una miotomía de Séller modificada con funduplicatura de Dor.
Comentarios: La acalasia es una entidad muy rara en niños, pero debe entrar dentro del diagnóstico diferencial de los vómitos y las infecciones respiratorias recurrentes. Muchos pacientes reciben tratamientos para otras enfermedades, como reflujo gastroesofágico, vómitos psicógenos o alteraciones del comportamiento alimentario, por mala interpretación de los datos, no por presentaciones atípicas. La radiología clásica muestra un esófago dilatado y en pico de pájaro distalmente. Debe realizarse una endoscopia para descartar causas secundarias de estenosis esofágicas. La manometría esofágica, con aumento del tono basal del esfínter esofágico inferior y no relajación tras la deglución, sugiere el diagnóstico.
P180 PÓLIPO JUVENIL INFLAMATORIO DE COLON COMO CAUSA DE RECTORRAGIA RECURRENTE EN UN PREESCOLAR DE 16 MESES. 09:10 h
María Rodríguez Martínez, Purificación Aguilera Sánchez, Encarnación López Ruzafa, Manuel Martín González, Francisco Morales Ferrer, Diego Vallejo Díaz, Salvador Fernández Dozagarat, Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
Antecedentes y objetivos: El pólipo juvenil de colon es el tumor intestinal más frecuente en la infancia.Suele localizarse en zona rectal o rectosigmoidea y son aislados en el 92,3%, con base de implantación pediculada.Con tamaño desde unos mm hasta los 3 cm. Escaso riesgo de malignización. La etiología es desconocida. Su incidencia varía desde el 0,61 al 1%, siendo la edad más frecuente de aparición entre los 2 y 8 años. No existe predominio en cuanto al sexo. Se presenta el caso de una niña con cuadro clínico consistente en episodios recurrentes de rectorragias con intervalos de deposiciones normales de edad atípica.
Métodos y resultados: Lactante mujer de 16 meses, con cuadro de 2 meses de evolución de episodios recurrentes de deposiciones con hebras de sangre roja mezclada con moco (en 5-6 ocasiones) con intervalos de 1-2 semanas de deposiciones normales, sin dolor abdominal ni fiebre, hábito intestinal normal y apetito conservado. Peso 10 Kg y talla 77 cm, exploración general, abdominal y tacto rectal sin datos relevantes. Hemograma, fórmula y recuento leucocitarios, bioquímica hepática y renal e Ig normales, serología de celiaquía, IgE específica a proteínas de leche de vaca y coprocultivos negativos. Las pruebas de imagen incluyeron Eco abdominal que informa de masa de 2,5 cm en ángulo esplénico de colon, existen dudas de si es intraluminal o no, una Eco posterior informa de masa de 2,5 cm en hipocondrio izquierdo que produce imagen de invaginación probable de intestino delgado. Ante discordancia de información se programa tránsito intestinal: defecto de replección solitario en ángulo esplénico, de localización colonica intraluminal, pediculada. Enema opaco no concluyente por dificultades técnicas. Colonoscopia: pólipo de gran tamaño, base sesil, aspecto inflamatorio en región esplénica de colon, se toman 3 biopsias, que informan de tejido de granulación de características inflamatorias. Ante hallazgos endoscópicos y anatomopatológicos se realiza cirugía (polipectomía transabdominal) por el gran tamaño y localización del pólipo.
Conclusiones: Se destaca su presentación en una edad no habitual, así como la localización y las características macroscópicas infrecuentes. A pesar de su rareza debe formar parte del diagnóstico diferencial de una rectorragia en edad preescolar.
P181 ESPASMO PILÓRICO CON PÁNCREAS ECTÓPICO ANTRAL Y OBSTRUCCIÓN GÁSTRICA COMO CAUSA DE DOLOR ABDOMINAL. 09:15 h
Aida Hernández Blanco, José M. Barroso Jornet, Albert Feliu Rovira, Gemma Castillejo de Villasante
Hospital Universitari de Sant Joan Societat Anònima Municipal, Reus (Tarragona) y Universistat Rovira i Virgili, Reus (Tarragona).
Introducción: Páncreas ectópico es una rara entidad que habitualmente suele ser un hallazgo incidental. La mayoría de los pacientes con páncreas ectópico son asintomáticos, y si tienen síntomas, éstos son inespecíficos y acordes según el lugar de la lesión. Su localización más frecuente es el antro del estómago y una rara presentación es la obstrucción gástrica.
Caso clínico: Niña de 10 años es traída a urgencias por dolor cólico abdominal en flanco izquierdo de manera súbita. Refiere dispepsia y sensación de plenitud acompañada de abdominalgia leve de predominio postpandrial semanas antes. No refiere vómitos ni otra sintomatología. Hábitos deposicionales normales. Personalidad nerviosa sin otros antecedentes de interés. Exploración física: dolor a la palpación periumbilical y en flanco izquierdo sin signos de irritación peritoneal. Radiografía de abdomen muestra desplazamiento inferior del colon transverso con imagen sugestiva de aumento del tamaño de cámara gástrica que se encuentra poco aireada. Analítica sanguínea: Hto 36,4%, leucocitos 6.700/mm3, amilasa 0,52 uKat/L, GOT 0,53 uKat/L, urea 6 mmol/L, creatinina 45 umol/L. El dolor cede a las pocas horas, con buena tolerancia alimentaria, pero reincide con mayor intensidad al día siguiente, objetivándose bazuqueo gástrico a la exploración abdominal. Tras aspirar 600 ml de contenido gástrico mediante SNG y cese del dolor inmediato, se realizan las siguientes pruebas: ecografía abdominal que objetiva engrosamiento de la mucosa del antro gástrico de 9 mm. TGED normal. Fibrogastroscopia: en el antro prepilórico se observa una pequeña formación polipoidea umbilicada en su centro de 8-10 mm compatible macroscópicamente con páncreas ectópico anular. Tras seguimiento por Gastroenterología Pediátrica no ha vuelto a presentar cuadros sugestivos de oclusión gástrica.
Conclusión: Páncreas ectópico es la anomalía congénita más frecuente en el antro del estómago. En algunos pacientes hay una tendencia a producir dolor abdominal intermitente. El efecto del tejido ectópico puede causar dismotilidad y/o espasmo local, en este caso pilórico, que añadido a factores psicosomáticos pueden acentuar dicho espasmo. El resultado puede ser una obstrucción intermitente del estómago. El páncreas ectópico anular debería ser considerado dentro del diagnóstico diferencial de la distensión o dilatación gástrica aguda.
P182 PANCREATITIS AGUDA POR LITIASIS BILIAR EN EDAD PEDIÁTRICA. 09:20 h
Gonzalo González García, Elena Muñoz Jalle, Mercedes Gracia Casanova, Gloria Bueno Lozano, Aurora Lázaro Almarza, Matilde Viñas Viña
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción: La pancreatitis aguda es una enfermedad con baja incidencia en la edad pediátrica (1/50.000) pero de gran importancia por su curso clínico impredecible y mortal en ocasiones. Sus causas son muy variadas, siendo las más frecuentes en la infancia: los traumatismos abdominales, las infecciones virales y la litiasis biliar.
Caso clínico: Niña de 13 años de edad, afebril, que tras trasgresión dietética presenta vómitos y epigastralgia progresiva. Exploración: Peso 79 Kg, talla 161 cm, IMC 30, regular estado general, abdomen con dolor difuso a la palpación superficial en epigastrio e hipocondrio derecho con hepatomegalia de 2 traveses de dedo, peristaltismo conservado y sin signos de irritación peritoneal. Pruebas complementarias: Leucocitos 23,9 mil/mm3 (86% de neutrófilos), glucemia de 190 mg/dl, perfil lipídico normal, LDH 738 UI/L, AST 191 U/L, ALT 515 U/L, GGT 209 U/L, bilirrubina total 4,3 mg/dl, amilasa sérica 1845 UI/L, amilasa en orina 10891, lipasa pancreática 946 UI/L. Ecografía abdominal: abundante líquido peritoneal libre. TAC abdominal: aumento de tamaño de cabeza y cola de páncreas, con áreas hipodensas, desestructuración completa del cuerpo de páncreas, apreciando a dicho nivel sensación de masa inflamatoria hipodensa, que se extiende hacia la transcavidad de los epiplones, mesocolon transverso, mesenterio y espacio perirrenal anterior izquierdo (probable flemón). Tratamiento: reposo digestivo, sonda nasogástrica de aspiración continua, rehidratación i.v., analgesia parenteral, omeprazol i.v. y ciprofloxacino i.v. Al 5º día se instauró nutrición parenteral. Se hicieron controles seriados de glucemia y calcemia que fueron normales. Ecografía abdominal (7º día): abundante contenido hipoecoico en vesícula compatible con microlitiasis.TAC (7º día): mala definicíon del proceso corporo-caudal pancreático con borrosidad de los planos peripancreáticos y aumento de la densidad de la grasa peripancreática. La evolución clínica fue favorable y se dió el alta a los 19 días. Se programó en el servicio de cirugía una colecistectomía posterior.
Conclusiones:1) La pancreatitis aguda debe formar parte del diágnostico diferencial del dolor abdominal agudo en niños con edad escolar. 2) La presencia de IMC P > 97 debe alertar sobre la posibilidad de la litiasis biliar como causa de la afectación pancreática.
P183 ENFERMEDAD DE CROHN METASTÁSICA. 09:25 h
Vanessa Alonso Morales, M. Pilar Ranchal Pérez, Marta Cruz Cañete, Silvia Ortega Pérez, Javier Blasco Alonso, Juan David González Rodríguez, Alfredo Barco Gálvez, Luis del Río Mapelli, Carlos Sierra Salinas, Antonio Jurado Ortiz
Hospital Materno Infantil Carlos Haya, Málaga y Hospital Universitario Virgen del Rocío, Sevilla.
Introducción: La enfermedad de Crohn (EC) metastásica se define como la aparición de granulomas no caseificantes en zonas anatómicamente distantes del área gastrointestinal, separadas de esta por tejido sano, en el contexto de dicha enfermedad. La morfología de las lesiones cutáneas metastásicas es variable: úlceras, pápulas, placas o nódulos que se suelen localizar en miembros inferiores, vulva, pene, tronco o cara por orden de frecuencia.
Casos clínicos:1) Niño diagnosticado de EC ileocólica y perianal con 6 años de edad. Ha seguido tratamiento con mesalazina, prednisona, 6-mercaptopurina, infliximab y otros inmunosupresores. A los 9 años de edad aparecen lesiones cutáneas en pene y escroto que fueron diagnosticadas anatomopatológicamente como granulomas no caseificantes (Fotografía 1). Estas lesiones mejoraron con corticoides y metronidazol, sufriendo exacerbaciones al disminuir la dosis de corticoides. 2) Niña diagnosticada de EC con ileítis y pancolitis a los 10 años de edad. Ha seguido tratamiento con prednisona, mesalazina, azatioprina, así como infliximab por mala respuesta clínica. Desde el inicio del cuadro presenta lesión en labio mayor derecho (Fotografía 2), que mejora hasta casi desaparecer con los corticoides y empeora cuando lo hacen los síntomas digestivos. Esta lesión es compatible con un granuloma y por tanto, con un cuadro de EC metastásica.
Conclusión: La EC metastásica es la manifestación cutánea menos frecuente de la EC. Suele presentarse en pacientes ya diagnosticados (Caso 1), aunque se han descrito casos de afectación simultánea (Caso 2) e incluso que precedieron a la clínica digestiva. El conocimiento de las manifestaciones cutáneas de la EC es por ello, interesante y útil para su diagnóstico y seguimiento.
P184 REVISIÓN DE LOS HALLAZGOS ENDOSCÓPICOS EN 67 CASOS DE HEMORRAGIA DIGESTIVA ALTA EN PACIENTES PEDIÁTRICOS. 09:30 h
Patricia Pernas Gómez, M. Carmen García Barreiro, Celia M. Rodríguez Rodríguez, Clara García Cendón, Gemma Novoa Gómez, Susana Rey García, Cristina Lorenzo Legeren, Santiago Fernández Cebrián, Carlos García Rodríguez, Federico Martinón Sánchez
Complejo Hospitalario, Ourense.
Antecedentes y objetivos: La hemorragia digestiva alta (HDA), hematemesis de sangre fresca o en "poso de café", tiene una incidencia baja aunque no excepcional siendo la endoscopia digestiva alta (EDA) el procedimiento diagnóstico de elección que permite el tratamiento hemostático si es preciso. No obstante, sus indicaciones no están estandarizadas y la revisión de una nueva serie contribuirá a su delimitación.
Métodos: Análisis retrospectivo de 67 episodios de HDA, asociada o no a hemorragia digestiva baja (HDB), con edades entre un mes y 14 años a los que se practicó una EDA.
Resultados: De 581 EDA, 67 (12%) fueron HDA correspondiendo el 56,7% a niños y el 43,3% a niñas. La edad media fue 75,27 meses (IC 95% 62,9, 87,5). Fueron hematemesis el 62,7% y el 37,3% en "posos de café", coincidiendo HDA y HDB en el 10%. La edad media de los que tuvieron hematemesis fue significativamente mayor que los que presentaron "posos de café", 84,1 meses (IC 95% 39,8, 80,9) frente a 60,4 meses (IC 95% 39,8, 80,9). Se registró fármaco potencialmente lesivo en 7 (10,4%): 71.4% AAS (con reducción progresiva en su empleo), 14,3% ibuprofeno, 14,3% corticoides. Las lesiones endoscópicas más frecuentes: erosión superficial gástrica (22,4%), hiperemia gástrica (20,9%), RGE (17,9%), nodulación en cardias (16,4%), hiperemia duodenal (14,9%), hernia hiatal deslizante (13,4%), hiperemia en cardias (11,9%), sin evidencia de hallazgos responsables (22,4%). Histológicamente destacan: gastritis aguda (23,5%), esofagitis crónica (22,4%), gastritis crónica (20,9%), sin hallazgos (22,4%), no valorable (11,9%) y otros.
Conclusiones: En nuestros resultados destaca: ausencia de sangrado activo que requiriese hemostasia; baja incidencia de úlceras digestivas, posiblemente en relación con el uso de protectores gástricos; reducción progresiva de observaciones en que se administró AAS, relacionado con su menor empleo; y la ratificación de ausencia de lesiones demostrables en un porcentaje alto, concordancia entre observación endoscópica e histopatológica. En conjunto la selección de los pacientes ha sido correcta demostrándose la EDA como un examen seguro para el diagnóstico y guía de tratamiento con ausencia de complicaciones.
P185 PÁNCREAS DIVISUM ASOCIADO CON PANCREATITIS CRÓNICA. 09:35 h
M. Teresa Fernández Castaño, M. Carmen de Fuentes Acebes, José Manuel Marugán de Miguelsanz, J. Espinel Díez, M. Blanca Herrero Mendoza, M. Belén Robles García
Hospital de Léon, León.
Introducción: El páncreas divisum es la anomalía congénita más frecuente del páncreas. Su papel como causa de pancreatitis aguda y crónica, y dolor abdominal recurrente, es controvertido. Aunque la anomalía anatómica está presente desde el nacimiento, muchos casos son asintomáticos hasta la edad adulta. La experiencia clínica en niños es escasa y poco clara.
Caso clínico: Paciente de 14 años afecta de síndrome de Norman, remitida por su pediatra por un cuadro de vómitos persistentes de unos 4 meses de evolución, con frecuencia biliosos tras las comidas y abdominalgia que irradia a espalda y zona lumbar derecha. Afebril, sin diarrea y con acolia ocasional. Cursa en episodios, con períodos intercríticos normales. Presenta un hemograma normal, en la bioquímica sérica, destaca una amilasemia de 861 U/L, amilasuria de 9882 UI/L, lipasa de 278, tripsina y perfil hepático completo normal.
Ecografía abdominal: imagen redondeada detrás del estómago y medial al duodeno, que podría corresponder a quiste de duplicación, divertículo duodenal o pseudoquiste pancreático.
Tránsito esófago-gastro-duodenal: Bulbo y marco duodenal sin alteraciones.
Colangio-resonancia: no aporta más información.
TAC abdominal: masa que ocupa la luz duodenal en tercio superior de duodeno descendente y que aparentemente se origina en la pared medial del mismo y parece existir un plano de clivaje que la separa de la cabeza pancreática. Sugiere mismas probabilidades diagnósticas que la ecografía.
Ante el hallazgo de lesión quística en la encrucijada bilio-duodeno-pancreática, de origen incierto, sin componente obstructivo duodenal, pero con episodios de pancreatitis agudas recurrentes, se decide realización de CPRE: páncreas ventral, vías biliares normales y se observa una rama pancreática que cruza completamente al duodeno y termina en una cavidad Diagnóstico: Páncreas divisum. Páncreas anular no obstructivo y cavidad pseudoquística comunicada con el conducto central.
Evolución: Tras evaluación quirúrgica se decide actitud expectante. La paciente sigue presentando crisis recurrentes y transitorias de pancreatitis, resueltas de forma conservadora, sin modificarse con enzimas pancreáticos con la ingesta. Se discute el manejo médico y/o quirúrgico del cuadro.
P186 DIARREA INTRATABLE DE LA INFANCIA SECUNDARIA A INTOLERANCIA A POLÍMEROS DE GLUCOSA. 09:40 h
Antonia Roca Jaume, Antonio Rosell Camps, Queralt Soler Campins, Miguel Fiol Jaume, Consuelo Galiana Ferre, Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca (Baleares).
Introducción: La diarrea intratable de la infancia se ha definido como un aumento de la excreción fecal diaria que se asocia habitualmente a un mayor contenido de agua en las heces, de severidad suficiente para requerir soporte nutricional, a menudo en forma de nutrición parenteral. La etiología subyacente es muy variada. Presentamos dos neonatos con un cuadro de diarrea intratable, debido a una colitis alérgica y que posteriormente desarrollaron intolerancia a polímeros de glucosa.
Casos clínicos: Los dos pacientes debutan en la primera semana de vida con un cuadro de distensión abdominal y deposiciones dispépticas abundantes. Presentan mejoría durante los períodos de ayuno y reaparición de la clínica con la introducción de alimentación con lactancia materna, fórmulas semielementales y fórmulas elementales. Ambos pacientes presentan distrofia progresiva, precisando nutrición parenteral exclusiva. Se realiza biopsia intestinal objetivándose infiltrado inflamatorio con predominio de eosinófilos. Uno de los pacientes presenta un Prick a proteinas de leche de vaca positivo, y el otro, test de hidrógeno espirado positivo tras sobrecarga de polímeros de glucosa. Tras la introducción de fórmula (3232-A. Mead-Johnsson) sin hidratos de carbono y con proteinas lácteas hidrolizadas, suplementándose con fructosa y posteriormente glucosa ambos pacientes presentan ganancia ponderoestatural importante con normalización de deposiciones. La evolución ha sido buena, tolerando dieta normal a los 2 y 21 meses respectivamente.
Comentarios: El tratamiento de la diarrea intratable se basa en nutrición parenteral y nutrición enteral con fórmula elemental. En algunos casos la nutrición enteral puede no ser tolerada, pudiendo corresponder una a intolerancia a los polímeros de glucosa que llevan las fórmulas elementales. El diagnóstico se realiza mediante test de hidrógeno espirado positivo con sobrecarga de polímeros de glucosa y negativo a la sobrecarga con glucosa y por la evolución clínica tras iniciar fórmula sin polímeros de glucosa. El tratamieto consiste en administrar una fórmula sin hidratos de carbono suplementandola con fructosa o glucosa. Su evolución es buena consiguiéndose tolerancia posteriormente.
P187 CARACTERÍSTICAS EPIDEMIOLÓGICAS DE LA GASTROENTERITIS AGUDA EN NUESTRO MEDIO EN LOS ÚLTIMOS 5 AÑOS. 09:45 h
María Garatea Rodríguez, M. Teresa Hernández Lagunas, Diana Martínez Cirauqui, Beatriz Solís Gómez, Elena Aznal Sáinz, Félix Sánchez-Valverde Visus
Hospital Virgen del Camino, Pamplona (Navarra).
Introducción: La gastroenteritis aguda (GEA) es uno de los diagnósticos mas frecuentes en los ingresos de pediatría (entre 8 y 12% de los ingresos en nuestro hospital en los 5 últimos años han sido gastroenteritis aguda). Revisamos en esta comunicación las características epidemiológicas de esta entidad clínica.
Material y métodos: Se revisan los pacientes ingresados en nuestro Servicio con el diagnóstico de GEA entre los años 1999 y 2003 y sus características epidemiológicas. Los diagnósticos al alta han sido recogidos sistemáticamente en nuestro servicio desde 1999 mediante una unidad de codificación diagnóstica. Los datos se han explotado en SPSS 11.0 para Windows, y se han analizado de cada registro las siguientes variables: edad, sexo, coprocultivo, duración del ingreso, enfermedad de base y complicaciones de la gastroenteritis.
Resultados: La duración media del ingreso fue de 2,88 días con un rango entre 1 y 37 días con una desviación típica de 2,4 días. La edad media al ingreso fue de 35 meses. Se analizaron los casos por grupos de edad encontrándose que el 78% de los niños eran menores de 4 años y el 57,8% de los casos eran menores de un año. El coprocultivo fue negativo en el 51% de los casos. Entre los positivos el 40% fue para rotavirus, (este porcentaje se eleva al 54% en el primer año de vida), el 34% para salmonella, el 16,5% campylobacter y el 2% para yersinia siendo el 7,5% positivo para otros microorganismos enteropatógenos. Se comprobó que de la gastroenteritis causada por rotavirus el 98,5% eran menores de 4 años y el 82% eran menores de 1 año. El 14,5% de los pacientes que ingresaron tenían una enfermedad de base y el 9,6% de los ingresaron tuvieron alguna complicación derivada de la gastroenteritis como deshidratación, acidosis o hipernatremia.
Conclusiones: La gastroenteritis aguda sigue siendo una patología prevalente como causa de ingreso en la edad pediátrica. El rotavirus es el enteropatógeno más habitual en el primer año de vida.
P188 HIDROPS VESICULAR AGUDO VERSUS COLECISTITIS AGUDA ALITIASICA EN NIÑO DE 7 AÑOS. 09:50 h
Juana Barja Tur, M. Luisa Herreros Fernández, Ana M. Pastor Gómez, Ricardo Díez García, Alfonso González Laguillo
Clínica Moncloa, Madrid.
Caso clínico: Escolar de 7 anos de edad que presenta fiebre alta (T max 39,5 C) de 4 días de evolución, acompañada de dolor abdominal epigástrico y nauseas. Coluria e hipocolia acompañante.
Antecedentes personales: Sin interés salvo 2 neumonías de LII, y dos crisis febriles típicas. Estancia de 2 meses en Ecuador, hasta una semana antes del inicio de los síntomas.
Antecedentes familiares: Madre diarrea prolongada durante su estancia en Ecuador.
Exploración física: Abdomen blando y depréciale, con dolor abdominal en hipocondrio dcho, con defensa a ese nivel y Murphy +, sin signos de irritación peritoneal. No ictericia. Resto de exploración sin hallazgos patológicos.
Exploraciones complementarias: Ecografía abdominal: marcada distensión de vesícula biliar con pared engrosada, y banda hipoecoica liquida perivesicular sin litiasis ni dilatación de via biliar.
Analítica de sangre: Hemograma, formula y recuento normales. Aumento de transaminasas y patrón colestasico.
Evolución clínica y tratamiento: tras su ingreso se inicia tratamiento conservador con dieta absoluta, sonda nasogastrica, analgesia y fluidoterapia y antibioterapia intravenosas.
Durante los primeros 4 días mantiene nauseas y dolor abdominal, acompañados de prurito marcado, precisando tratamiento con Resincolesteramina. Posteriormente evolución favorable con mejoría clínica y analítica hasta desaparición del dolor, con normalización ecografica a la semana del ingreso, con buena tolerancia oral.
Se confirma durante el ingreso infección aguda por VHA con negatividad de bacteriología y parásitos. Normalización analítica a las 2 semanas del alta.
Conclusiones:a) Tanto la colecistitis aguda alitiasica, como el hidrops vesicular agudo, son infrecuentes en la infancia, y de difícil diferenciación entre ambas, debiendo tenerse en cuenta en el dco diferencial del dolor abdominal agudo en el niño, b) En ambas patologías, secundarias con frecuencia a patología infecciosa en el niño, se ha demostrado efectivo el tratamiento conservador, con control clínico y eco gráfico, sin precisar en muchas ocasiones tratamiento quirúrgico.
INFECTOLOGÍA
P189 REVISIÓN DE INGRESOS HOSPITALARIOS POR VARICELA EN EL HOSPITAL UNIVERSITARIO DE VALME EN LOS ÚLTIMOS 10 AÑOS. 08:30 h
M. del Carmen Medina Gil, M. Ángeles Carrasco Azcona, Josefina Márquez Fernández, Laura Acosta Gordillo, Israel Coronilla de Luis, Rafael Espino Aguilar
Hospital Universitario de Valme, Sevilla.
Introducción: La varicela, aunque considerada propia de la infancia y "benigna", puede causar complicaciones e ingresos hospitalarios con el consiguiente coste. La reciente comercialización de una vacuna protectora frente a la varicela en España que podría evitar estas repercusiones, nos lleva a presentar esta revisión.
Método: Estudio retrospectivo sobre pacientes pediátricos con diagnóstico al alta de varicela en nuestro hospital en el período 1992-2002. Se analizaron edad, sexo, período estacional, antecedentes clínicos, causas de ingreso, días de evolución previos y de hospitalización, tratamientos previos y hospitalarios, exámenes complementarios e interconsultas realizadas, evolución y diagnóstico al alta.
Resultados: Se registraron 79 casos: 0,49% de los ingresos hospitalarios. Distribución por sexos: 1:1. Edad: entre recién nacidos y 14 años, con un pico entre los 2-5 años. Máxima incidencia de casos en verano con un máximo de ingresos en 1995. La mayoría sin antecedentes clínicos de interés. Causas de ingreso: destacan fiebre resistente a antitérmicos (45%) y convulsiones febriles (20%) frecuentemente previas a la aparición del exantema. La evolución media del exantema al ingreso fue 3 días y de 6 la hospitalización. 75% de casos no realizó tratamiento previo al ingreso. Exámenes complementarios más solicitados: hemograma, bioquímica, radiografía de tórax, orina y electroencefalograma. En 8% no se indicó ninguna prueba. Se prescribió: 65% aislamiento, 45% antitérmicos, 35% antibioterapia, 26% antihistamínicos, 22% aciclovir y 8% anticonvulsivantes. 10% de casos precisó sólo antitérmicos y aislamiento. El diagnóstico de varicela se asocia en 50% de casos a complicaciones, siendo las más frecuentes las sobreinfecciones (29%) cutáneas y respiratorias (15% y 14% respectivamente) seguidas de las neurológicas (25%).
Discusión: Del total de ingresos hospitalarios 0,49% fueron debidos a varicela. La estancia media hospitalaria, que conlleva el absentismo laboral de los familiares, las exploraciones complementarias y la medicación, suponen un coste económico y social importante. Destaca el alto porcentaje de complicaciones neurológicas. Se produjo un fallecimiento por neumonía varicelosa en un paciente inmunodeprimido.
Conclusión: Disponiendo de una vacuna eficaz frente a la varicela, cabría plantearse su administración de forma sistemática a la vista de los resultados.
P190 AVALIAÇÃO DE UM PROGRAMA PARA REDUZIR AS TAXAS DE TRANSMISSÃO VERTICAL DO HIV NO BRASIL - RESULTADOS DE UM ESTUDO COLABORATIVO MULTICÊNTRICO. 08:35 h
Regina Célia Succi, Suzane Cerutti Kummer
Grupo Estudo Soc. Brasileira Pediatría, Sociedade Brasileira de Pediatría, São Paulo (Brasil).
Antecedentes e objetivos: Casos de transmissão vertical (TV) do HIV são descritos no Brasil desde 1984, mas o número de crianças infectadas por essa via tem diminuído desde 1996. O Brasil oferece livre acesso a terapia antiretroviral para o tratamento de AIDS e desde 1995 a prevenção da TV do HIV é considerada prioridade. Estima-se que 0,6% das gestantes brasileiras estejam infectadas pelo HIV. O objetivo deste estudo colaborativo multicêntrico foi avaliar as taxas de TV do HIV em todas as regiões do país entre crianças nascidas no período compreendido entre janeiro de 2000 a dezembro de 2002.
Material e métodos: 63 serviços (localizados em 20 estados e no DF) participaram do estudo. 4004 crianças foram analisadas (1462 nascidas em 2000, 1462 em 2001 e 1080 em 2002). A TV do HIV nos três anos foi de 6,76% (271/4004), sendo 8,6% em 2000, 7,1% em 2001 e 3,8% em 2002. A TV do HIV foi maior nas regiões norte (10,4%) e nordeste do país (12,3%) quando comparada com as regiões centro-oeste (5,6%), sul (5,5%) e sudeste (6,8%). O diagnóstico da infecção pelo HIV foi feito antes ou durante a gestação em 86% das gestantes, mas entre aquelas transmissoras do vírus essa taxa foi de 52,0%, enquanto que para as não transmissoras foi de 88,6%. O diagnóstico apenas durante a internação hospitalar para o parto, realizado através de teste rápido para o HIV, ocorreu em 196 das 4004 mulheres estudadas (4,9%) e destas, 35 (17,8%) transmitiram o HIV para seus filhos. 80% das mães tiveram acesso à terapia antiretroviral na gestação e parto; 92% dos recém nascidos (RN) receberam AZT. Apenas 5,6% dos RN receberam aleitamento materno e nestes, a TV do HIV foi maior (33,3%) do que entre aqueles que receberam fórmula látea (5,0%). 40% das mulheres foram submetidas à cesárea eletiva (antes do trabalho de parto) e a TV do HIV nestes RN foi igual a 2,7% e 10,3% entre aqueles nascidos de parto vaginal.
Conclusões: A TV do HIV no Brasil vem diminuindo nos três anos avaliados em conseqüência da utilização de medidas preventivas. Nesse período foi possível verificar associação de maiores taxas de TV do HIV com aleitamento materno, parto vaginal, falha no uso da terapia antiretroviral profilática nas mães e no RN.
P191 ESTUDIO DESCRIPTIVO DE PACIENTES INGRESADOS POR BRONQUIOLITIS. 08:40 h
David Canalejo González, M. Esther García Rodríguez, Víctor Manuel Navas López, Elia Sánchez Valderrábanos, M. Teresa Charlo Molina
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivos: La bronquiolitis es una enfermedad epidémica potencialmente grave, con elevada morbimortalidad, que ocasiona un gran número de consultas e ingresos hospitalarios. Describir el manejo de los pacientes con bronquiolitis aguda en nuestro centro y sus características.
Material y método: Estudio descriptivo retrospectivo donde se incluyen 121 historias clínicas de pacientes ingresados en nuestro hospital desde Octubre de 2002 hasta Marzo de 2003 con diagnóstico al alta de bronquiolitis.
Resultados: Hubo un predominio en menores de 6 meses (85%), especialmente en neonatos. Ligero predominio de varones (56,2%). Casi el 50% de los pacientes eran alimentados con lactancia materna. La sintomatología respiratoria menor estaba presente en la mayoría de los casos. Otros síntomas presentes fueron fiebre y febrícula (57,8%), rechazo del alimento (47,1%) e irritabilidad (6,6%). Los motivos de ingreso más frecuentes fueron el distress moderado/apneas y la imposibilidad de mantener una hidratación adecuada. Un 47% fue catalogado como pacientes de alto riesgo. La radiografía de tórax presentaba imágenes de atrapamiento aéreo (42,2%), atelectasias laminares (18,2%), condensación (19%) y aumento de la trama bronquial (43,8%). El hemograma mostraba una linfomonocitosis total o relativa (70,2%) y la gasometría una acidosis respiratoria (51,6%). La detección del VRS fue positiva en el 53,9%. El 97% de los pacientes monitorizados presentaba una llamativa repercusión oximétrica. El 63% de los pacientes presentaba una puntuación en el Score de Downes entre 4 y 7, y el 8% presentaba un Score mayor de 7. El tratamiento recibido fue salbutamol inhalado (80,1%), corticoides orales (77,7%) o anticolinérgicos inhalados (62,8%), generalmente combinados. La adrenalina nebulizada se utilizó en el 29,7% de los pacientes. El rango de días de estancia hospitalaria osciló entre 1 y 23 días, con una mediana de 6 días. El 10% de los pacientes precisó ingreso en UCI. La mortalidad fue del 0,8%.
Conclusiones: La bronquiolitis es una enfermedad potencialmente grave. La pulsioximetría y el Score de Downes son una ayuda fundamental para indicación de tratamiento hospitalario. Existe actualmente la necesidad de crear un protocolo que unifique la actuación diagnóstico y terapéutica del médico.
P192 A PROPÓSITO DE UN CASO DE TROMBOPENIA SEVERA AGUDA SECUNDARIA A VARICELA. 08:45 h
Carmen Fresneda Machado, José M. López Gómez, Vicente J. Ramírez Piedrabuena, M. Teresa Moya Díaz-Pintado, Mª Sierra Antona García, Arturo Sánchez Enfedaque
Hospital Virgen de Altagracia de Manzanares, Ciudad Real.
Se presenta el caso de un niño de 9 años sin antecedentes personales ni familiares de interés que presenta hematuria franca y lesiones cutáneas purpúricas en el contexto de una varicela de 3 días de evolución.
Exploración: Afebril, hemodinámicamente estable. Erupción cutánea generalizada con lesiones en cielo estrellado, vesículas hemorrágicas, sangrado agudo de las lesiones en mucosa geniana, hematuria macroscópica.
Resto de exploración normal.
Pruebas complementarias:a) Hemograma: plaquetas 1000; resto normal, b) Coagulación: normal, c) Rx de tórax: normal.
Tratamiento: Se administra aciclovir i.v. a 50 mg/kg/día, gammaglobulina i.v. a 0,8 g/kg i.v. y se transfunden 3 unidades de plaquetas remontando la trombopenia y desapareciendo la cínica de diátesis hemorrágica.
Evolución: Quince días después presenta trombopenia de 48000 plaquetas sin clínica acompañante por lo que se inicia estudio de púrpura trombopénica idiopática.
Conculsiones: Se trata de un caso de varicela complicada con trombopenia severa en un paciente inmunocompetente. Esta complicación es rara y puede tratarse de una trombopenia transitoria aislada o desarrollar una púrpura trombopénica idiopática secunaria a la infección viral por el virus varicela zoster. La varicela es una enfermedad frecuente que ha sido considerada benigna en los niños, no así sus complicaciones que presentan una elevada morbilidad. Las complicaciones hematológicas son de las menos frecuentes pero en los casos de trombopenia severa como nuestro caso supone un riesgo vital. Por tanto sería beneficiosa la implantación de la vacuna de la varicela para prevenir sus complicaciones.
P193 ULCUS VULVAE ACCUTUM O ENFERMEDAD DE LIPSCHÜTZ. 08:50 h
Rocío Risquete García, Susana Cora López, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio, José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Objetivo: Comunicar una entidad clínica poco frecuente en la adolescente joven.
Material y método: Adolescente de 13 años de edad que presenta úlcera dolorosa en labio mayor izquierdo de dos días de evolución. Desde 24 horas antes presenta fiebre, deposiciones de menor consistencia sin restos patológicos y vómitos alimenticios. Niega relaciones sexuales. Menarquia hace 15 meses. A la exploración, buen estado general, piel y mucosas bien hidratadas y perfundidas, abdomen blando y depresible, peristáltica audible. Orofaringe hiperémica con exudado puntiforme en amígdala izquierda sin lesiones aftosas. Genitales femeninos bien configurados, úlcera en labio mayor izquierdo con borde inflamatorio y base necrótica de consistencia dura, inflamación perilesional, no adenopatias palpables. Hemograma: 11.900 leucocitos con 75,6% de neutrófilos; PCR: 84,2 mg/l; orina: normal; serología a sífilis, herpes virus, mononucleosis infecciosa, fiebre tifoidea y paratifoidea negativa; frotis cutáneo y exudado de úlcera: flora de origen cutáneo; aspirado cutáneo: cultivo negativo; informe oftalmológico: normal; mantoux: negativo. Tras descartar enfermedades venéreas, tuberculosis, enfermedad de Crohn y Behcet establecemos el juicio clínico de ulcus vulvae accutum o enfermedad de Lipschütz.
Resultado: Instauramos tratamiento con antibioterapia sistémica de amplio espectro y antisépticos locales siendo la evolución favorable, desapareciendo la fiebre y el dolor el segundo día tras su ingreso. Al alta, una semana después, no había signos inflamatorios y la úlcera se encontraba en proceso de resolución.
Discusión: La enfermedad de Lipschütz es un proceso no bien conocido y quizás por ello infradiagnosticado que se presenta en adolescentes jóvenes (7-25 años) que no han mantenido aún relaciones sexuales en su mayoría. Su etiología y patogenia no están aún bien establecidas aunque se sugiere la posibilidad de origen infeccioso. Lipschütz asume que es causada por la inoculación de lactobacilos de Döderlein aunque otros autores la atribuyen a mala higiene de la adolescente. En algunos casos se han identificado virus de Epstein-Barr y Ureaplasma. Úlceras muy similares se han encontrado en mujeres VIH positivas. También se ha asociado a fiebre tifoidea y paratifoidea. Se describen tres formas clínicas: gangrenosa, forma más frecuente y que describimos en nuestro caso, crónica y miliar.
P194 NEUMONÍAS DE REPETICIÓN POR CUERPO EXTRAÑO. 08:55 h
Elena Muñoz Jalle, Gonzalo González García, Mercedes Gracia Casanova, Xenia Alonso Curcó, A. Pérez Trullen
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción: La aspiración de un cuerpo extraño, es la causa más frecuente de muerte por accidente doméstico en niños menores de seis años. Es más frecuente en varones con dos picos de incidencia, a los tres y a los once años de edad.La clínica inicial consiste en un episodio de sofocación, y si el diagnóstico se difiere, se manifiesta como tos persistente, sibilancias y neumonías de repetición. La mortalidad en nuestro medio es del 0,9%.
Caso clínico: Varón de 14 años, sin antecendes patológicos previos, que presenta tos persistente desde hace diez meses. Diagnosticado en tres ocasiones de neumonía basal derecha y de asma intrínseco que no responde al tratamiento. Exploración: Fiebre de 38,5, no signos de dificultad respiratoria, auscultación pulmonar con disminución de murmullo vesicular en plano anterior y lateral de hemitórax derecho, y roce pleural posterior. Rx. de tórax: Neumonía basal derecha con derrame pleural. Tratamiento: Cefalosporina de 3º generación y corticoide con mejoría clínica y radiológica. Por sus antecendentes se insiste en la historia clínica, y refiere un episodio de atragantamiento con sensación de ahogo hace un año, al comer pipas. T.A.C. torácico: imagen compatible con cuerpo extraño a dos centímetros de bronquio lobar derecho.Se realizó broncoscopia que permitó extraer el cuerpo extraño y la visualización de un granuloma reactivo en dicha localización. Se pautó tratamiento con corticoide y se citó para una nueva broncoscopia a los dos meses en la que persistía el granuloma.En el momento actual pemanece asintomático.
Comentarios:1) Destacar la importancia de la historia clinica ante una sintomatología sugerente de aspiración, ya que muchos pacientes olvidaron el episodio de atragantamiento o no le dieron la suficiente importancia. 2) Las características del material aspirado son distintas dependiendo del grupo de edad; los frutos secos son la principal causa en los más pequeños y los objetos de uso escolar en los segundos. 3) La broncoscopia es el principal metodo diagnóstico y terapéutico, y esta indicado aun cuando la radiografía sea normal.
P195 GRANULOMATOSIS HEPATO-ESPLÉNICA COMO MANIFESTACIÓN ATÍPICA DE INFECCIÓN POR BARTONELLA HENSELAE. 09:00 h
Amagoia Andrés Olaizola, Rocío Lamarca Gay, Eider Astobiza Beobide, Itziar Pocheville Gurutzeta, J. Miguel Arana Herrerías
Hospital de Cruces, Barakaldo (Vizcaya).
Introducción: La enfermedad por arañazo de gato típica se caracteriza por la aparición de adenopatías regionales tras sufrir el arañazo de un gato. El agente causal es Bartonella henselae. Una forma atípica de la bartonellosis es la granulomatosis hepato-esplénica aislada, que no parece relacionada con el estado inmunológico del paciente.
Caso clínico: Niña de 9 años de edad, sin antecedentes personales de interés, salvo contacto con gatos y perros (entorno rural), que acudió a Urgencias por dolor abdominal constante, de 10 horas de evolución y fiebre de 38,5 º C. A la exploración presentaba dolor abdominal a la palpación fundamentalmente en fosa ilíaca derecha. Ante la sospecha de abdomen agudo, se realizaron analítica sanguínea y radiografías de tórax y abdomen, que fueron normales, y una ecografía abdominal que mostró 4 lesiones focales esplénicas (la mayor de unos 12 mm) con halo ecógenico y centro anecoico, de aspecto necrótico, sin objetivar apendicitis. En la TAC abdominal se observaron múltiples lesiones focales esplénicas menores de 1 cm, con centro hipodenso, y múltiples lesiones focales hepáticas mínimamente hipodensas respecto al parénquima, con cierto halo de hiperatenuación, sugestivas de granulomatosis. El Mantoux fue negativo. Las serologías realizadas resultaron negativas, excepto a Bartonella henselae, con aumento de los títulos 8 días más tarde. Se instauró tratamiento con cotrimoxazol y rifampicina vía oral durante 15 días. En posteriores controles, los títulos de anticuerpos fueron disminuyendo al igual que el número y tamaño de las lesiones hepato-esplénicas en el control ecográfico. Permaneció asintomática en todo momento.
Comentarios: Ante una fiebre de origen desconocido, con historia de exposición a gatos y dolor abdominal, aún en ausencia de la clínica típica de pápula roja y adenopatías (en esta forma se observan en un 60% y 50% respectivamente), debe descartarse una forma hepato-esplénica por enfermedad por arañazo de gato. Aunque este cuadro parece tener una evolución benigna, algunos autores recomiendan instaurar tratamiento antibiótico empírico (trimetoprim-sulfometoxazol, rifampicina, ciprofloxacino, o gentamicina sulfato) en espera de resultados de las serologías.
P196 UN CASO DE EPISODIO APLÁSICO PROVOCADO POR EL PARVOVIRUS B19. 09:05 h
Pilar Rojas Feria, María José Martínez Roda, Estrella Peromingo Matute, M. Soledad Camacho Lovillo, María Bueno Delgado, Juan Antonio León Leal
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción: El parvovirus B19 es el único miembro de la familia parvoviridiae que produce patogenicidad en humanos. La infección por este virus es muy común en la infancia. A la edad de 15 años casi el 50% tienen valores detectables de IgG.
Caso clínico: Niña de 6 años que consulta por episodios repetidos de epistaxis de 2 días de evolución. En las últimas 24 horas, lesiones petequiales puntiformes en MMSS e II y equimóticas en MMII. Cinco días antes presentó cuadro febril de 2 días de duración. Exploración al ingreso: B.E.G. Afebril. No signos meníngeos. Lesiones en miembros ya comentadas. Adenopatías de pequeño tamaño laterocervical izqda. ACP: Normal. Abdomen blando, depresible con hepatomegalia y mínima esplenomegalia. ORL: bulla hemorrágica en lengua. Resto sin hallazgos. Pruebas complementarias y evolución: Hg y frotis al ingreso: leucocitosis con linfomonocitosis (30% de linfocitos activados) y neutropenia leve, trombopenia, anemia microcítica hipocrómica. Coagulación: normal. A su ingreso se trata con gammaglobulina IV a 400 mg/Kg 5 días. A pesar de ello, continua con epistaxis repetidas. La persistencia de estos datos clínicos se corresponde con una trombopenia mantenida en el hemograma. A su vez se acentúa la neutropenia (830), y desciende la hemoglobina y el hto (85 y 25%), confirmándose la afectación de las tres series en sangre periférica. GOT 260 UI/l, GPT 356 UI/l, LDH 1017 UI/l, PA 530 UI/l. Resto normal. Metabolismo del hierro: sideremia 18 g/dl, ferritina 10 g/l, transferrina 311 mg/dl, I.S. 9%.VSG: 95 mm/h, PCR: 3.08 mg/l.Proteinograma, orina, Rx tórax: Normales. Ecografía abdominal: discreta esplenomegalia homogénea. Ligero aumento del tamaño del hígado. Autoanticuerpos: negativos. Médula ósea (M.O.): reactiva con trombopenia megacariocítica y alteraciones madurativas de las demás series. Subpoblaciones linfocitarias: sugestivas de proceso viral agudo. No blastos. Serología: parvovirus B19 IgM positivo. Resto negativo.
Discusión: El parvovirus B19 tiene un marcado tropismo sobre la M.O. humana. Las manifestaciones hematológicas tras su infección son debidas a la agresión de la célula precursora eritroide mediante un mecanismo citotóxico. Puede presentarse en algunos enfermos hematológicos sin producir clínica, y a veces, se llega al diagnóstico de una anemia de base, antes desconocida, tras la presentación de un episodio aplásico (como es el caso que nos ocupa).
P197 MENINGITIS TUBERCULOSA EN LACTANTE DE 18 MESES. 09:10 h
Ana Mata Fernández, Mercedes Cortés Martín, Julia Cano Fernández, M. José Cerezo Bueno, Enma de la Torre Montes de Neira, Gladys Yep Chullen
Hospital del Niño Jesús, Madrid.
Introducción: La meningitis tuberculosa afecta aproximadamente al 1% de los niños con enfermedad tuberculosa, siendo la complicación más grave de esta infección, con un pico de máxima incidencia entre 6 meses y 6 años. Una de las manifestaciones de la tuberculosis del Sistema Nervioso Central es el tuberculoma, de localización infratentorial en los niños, a diferencia de la localización supratentorial en los adultos.
Caso clínico: Lactante de 18 meses diagnosticado hace 3 meses en su hospital de referencia de tuberculosis pulmonar en tratamiento con isoniacida, rifampicina y piracinamida. Presenta desde hace 3 semanas ptosis palpebral izquierda, paresia de miembro inferior derecho, ataxia, decaimiento y somnolencia.
Pruebas complementarias: TAC craneal / RMN craneal: lesión focal con realces anulares en bulbo medular, con realces leptomeníngeos en las cisternas basales. TAC torácico: infiltrado en lóbulo superior izquierdo, con imágenes nodulares en parénquima unidas íntimamente a pleura. LCR: 48 células/mm3, con predominio de mononucleares, glucosa 55 mg/dl, proteínas 96 mg/dl, ADA 4,9 UI/l. tinción de Ziehl negativo, cultivo en medio de Lowestein negativo, PCR para M.tuberculosis negativo. Fondo de ojo: normal. Se instaura tratamiento tuberculostático con isoniacida, rifampicina, piracinamida, estreptomicina y corticoides. El paciente presenta una evolución favorable con mejoría de los síntomas neurológicos.
Comentarios: El diagnóstico de sospecha de meningitis tuberculosa justifica la instauración de tratamiento específico, con la finalidad de reducir las complicaciones y mortalidad de dicha enfermedad. El pronóstico de la meningitis tuberculosa depende principalmente de la edad, siendo peor en lactantes pequeños que en niños mayores, y del estadío de la enfermedad en que se inicie el tratamiento. La aparición de tuberculomas en un paciente con meningitis tuberculosa que recibe tratamiento tuberculostático está documentada, sin que signifique el fracaso de dicho tratamiento.
P198 PLEURONEUMONÍAS EN LA INFANCIA. REVISIÓN DE 27 CASOS. (2002-2003). 09:15 h
Nayra Dopazo Ramos, Zenaida Galve Pradel, Margarita Bouthelier Moreno, Carmen Marín Bravo, Fernando de Juan Martín
Hospital Materno Infantil Miguel Servet, Zaragoza.
Introducción: Dado el incremento actual de la prevalencia de neumonías complicadas en nuestro medio, hemos revisado los casos de pleuronemonías registrados en nuestro servicio en los últimos dos años.
Material y métodos: Revisión retrospectiva de 27 casos de neumonías con derrame pleural ingresados en la Sección de Infecciosos del Hospital Infantil "Miguel Servet" de Zaragoza durante los años 2002 y 2003.
Resultados: -Datos epidemiológicos.- Sexo: 17 varones (63%), 10 mujeres (37%); Período estacional: primavera 6 casos (22%), verano 3 (11%), otoño 7 (26%), invierno 11 (41%); Edad: 15 entre 2 y 5 años (55%), 5 en < 2 años (19%), 4 en > 10 años (15%), 3 entre 6 y 10 años (11%). Clínica.- Localización: derecha 16 (60%), izquierda 9 (33%), bilateral 2 (7%); Síntomas de inicio: fiebre en 24 (88,9%), tos en 14 (51,8%), abdominalgia en 6 (22,2%), dolor torácico en 6 (22,2%), distres en 5 casos (18,5%), vómitos en 5 (18,5%), CVA en 4 (14,8%). Laboratorio.- leucocitos en sangre < 5.000/mm3 en 1 (4%), entre 5-10.000 en 7 (26%), entre 10-15.000 en 8 (30%), entre 15-20.000 en 2 (7%) y > 20.000 en 9 (33%); neutrofilia > 70% en 19 (70%); trombocitosis > 400.000/mm3 en 7 casos (25,9%); VSG entre 15-50 mm en 5 (18,5%), > 50 mm en 19 (70,4%) y desconocida en 3 (11,1%); Ag neumocócico en orina en 9 (33%); cultivo liquido pleural positivo en 3 casos (11%), 2 S. pneumoniae y 1 S. pyogenes, negativo en 8 (30%) y no realizado en 16 (59%); hemocultivo positivo en 3 (11%): 2 a S. pyogenes y 1 a S. pneumoniae, no coincidentes con hemocultivo positivo; aspirado nasofaringeo:VRS en 4 casos (14,8%) y adenovirus en 3 (11,1%) y serología a M. pneumoniae en 4 (14,8%). Tratamiento.- Además de antibioterapia con cefotaxima o cefotaxima/vancomicina se realizó punción evacuadora en 3 casos (11,1%), drenaje en 8 (29,6%), fibrinolíticos en 1 (3,7%) y decorticación en 1 (3,7%).
Conclusiones: Predominio en varones. Mayor incidencia en invierno y otoño. Mas frecuente entre 2-5 años de edad. Localización predominante lado derecho. Al ingreso se registró leucocitosis > 20000 (33%) y neutrofilia (70%). La etiología bacteriana se desmostró en el 22,2%. Hubo necesidad de punción o drenaje en el 40% de los pacientes y la decorticación fue realizada en 1 caso.
P199 MENINGITIS EN LACTANTE PEQUEÑO POR S. AGALACTIAE DE EVOLUCIÓN PROLONGADA. 09:20 h
Ana Gutiérrez Amorós, Óscar Manrique Moral, Ángela Sempere Pérez, Ana Fernández Bernal, Esther Blanco Alemany, Olga Gómez Pérez, Juan Utrero Valiente, M. del Carmen Vicent Castelló, Amelia Herrero Galiana, José Flores Serrano
Hospital General Universitario, Alicante.
Introducción: La meningitis por S. agalactiae es propia del neonato y lactante pequeño. Es más grave, con mayor morbilidad (30-50%) y mortalidad (17-40%) que otras meningitis. Sin complicaciones la clínica en la meningitis suele ceder en 5 días, en el 10% la fiebre persiste más de 7 días, indicando complicaciones. El LCR es fundamental en el diagnóstico y las complicaciones pueden requerir punciones de repetición, que han de interpretarse junto al resto de datos clínico.
Caso clínico: Lactante de 30 días con fiebre, quejido y rechazo del alimento. RNT, S. agalactiae desconocido con profilaxis completa, parto eutócico.
Exploración: Mal estado general, mala perfusión, fontanela amplia, "llena", taquipnea superficial con quejido, hipotonía generalizada, y crisis convulsiva.
LCR con pleocitosis e hiperproteinorraquia, hemocultivo y cultivo LCR positivos para S. Agalactiae sensible a penicilinas y cefalosporinas. Tratada inicialmente con ampicilina y cefotaxima (2 días), cefotaxima (21 días) y posteriormente ampicilina a dosis altas (28 días). Lenta mejoría clínica y analitica, persistiendo fiebre (44 días) reactantes de fase aguda (40 días) y alteración celular y bioquímica del LCR. La ECO, TAC y RNM detectan leve dilatación ventricular. La búsqueda de otros focos (exploracion, analíticas y pruebas de imagen) resulta negativa. Tras una semana afebril, con normalidad analítica, se retira antibioticos (52 días totales) En ese momento el LCR presenta 110 leucos, 225 mg/dl proteínas y 30 mg/dl glucosa. En la actualidad el protocolo de seguimiento no detecta secuelas.
Discusión: Fiebre, ↑PCR y LCR muy alterado durante 1,5 meses, los datos citados generan una caso muy especial que plantea diagnóstico diferencial entre: complicaciones intracraneales, extracraneales, fiebre por drogas, fracaso terapeútico o una evolución lenta de la enfermedad. Las pruebas descartan focos a otro nivel y apuntan a persistencia inflamatoria a nivel de SNC. La ausencia de otras complicaciones en el estudio y, a pesar de la negativización de los cultivos con ausencia de imágenes claras salvo una leve dilatación ventricular, sugiere que podría tratarse de cuadro de ventriculitis atípica con respuesta a antibioterapia i.v. La presencia de pleocitosis al final del tratamiento, en ausencia de manifestaciones clínicas, no es indicativa de complicaciones ni prolongar la antibioterapia. A pesar de la lenta evolución llama la atención la ausencia de secuelas 6 m después.
P200 NECESIDAD DE REVALORACIÓN DE LOS CRITERIOS DIAGNÓSTICOS DE TUBERCULOSIS EN LA EDAD PEDIÁTRICA. 09:25 h
Ana Morillo Palomo, Victoria Fumadó Pérez, Giorgia Sebastiani
Hospital San Joan de Deu, Barcelona.
El objetivo de este estudio es describir cuatro casos clínicos de niños inmigrantes visitados en consultas externas de nuestro hospital que inicialmente no cumplían los criterios diagnósticos actuales de infección tuberculosa y que sin embargo fueron diagnosticados de tuberculosis pulmonar. A pesar de los avances técnicos de los que disponemos en la actualidad el diagnóstico de la tuberculosis en la infancia continúa siendo un gran desafío. En los últimos años la tuberculosis se ha convertido en una enfermedad emergente debido al aumento cada vez más frecuente de población inmigrante procedente de países con alta prevalencia y endemia de esta enfermedad y con mayores resistencias a los tratamientos utilizados para erradicar la enfermedad. En este estudio describimos cuatro casos de niños adoptados, todos ellos menores de 2 años de edad, que tenían un PPD inferior a 15 mm y radiografías sin alteraciones aparentes. Los cuatro presentaban escara de la BCG. Se les realizó un TAC pulmonar por sospecha clínica de infección hallándose en el primero una adenopatía calcificada, en el segundo un mazacote adenopático, en el tercero adherencias pleurales y un infiltrado en lóbulo medio y en el cuarto un infiltrado retrocardíaco. A todos ellos se les pautó tratamiento con triple terapia con la sospecha diagnóstica de enfermedad tuberculosa. Los criterios diagnósticos utilizados actualmente para el diagnóstico de la enfermedad tuberculosa en la edad pediátrica deben ser revisados debido al aumento de la prevalencia de esta patología en nuestra sociedad que conlleva una importante repercusión sobre todo en los pacientes pediátricos de menor edad.
P201 INGRESOS HOSPITALARIOS POR VARICELA (1996-2003). 09:30 h
M. Pilar Ranchal Pérez, David Moreno Pérez, Mª José García Arias, Olga M. Escobosa Sánchez, Vanessa Alonso Morales, Yolanda María Chica Fuentes, José Manuel Jiménez Hinojosa, Antonio Madrid Madrid, Francisco Jesús García Martín, Antonio Jurado Ortiz
Hospital Materno Infantil Carlos Haya, Málaga.
La varicela es una enfermedad exantemática cuyo espectro clínico varía desde un cuadro banal hasta una enfermedad grave con afectación importante del estado general y diversas complicaciones (cutáneas, respiratorias, neurológicas...) que suelen ser el motivo de ingreso, salvo casos especiales como el período neonatal o pacientes inmunodeprimidos.
Objetivos: Conocer el perfil de los pacientes que precisaron ingreso por varicela.
Método: Estudio retrospectivo de las hospitalizaciones relacionadas con varicela del 1 de enero de 1996 al 31 de diciembre de 2003. El diagnóstico de varicela fue clínico.
Resultados: 114 casos de varicela precisaron ingreso. Se observa un incremento anual, con dos picos máximos en 1999 (24 casos) y 2003 (27 casos). La media de edad fue de 4.1 años, con 23 casos menores de 1 año. No hay diferencias en cuanto al sexo (47,3% mujeres y 52,6% varones). La mayoría de los ingresos se produjeron entre marzo y julio (67%). De los 114 casos, 17 (15%) eran inmunodeprimidos, siendo el tratamiento inmunosupresor y la leucemia linfoblástica los motivos más frecuentes. Las complicaciones cutáneas (impetiginización y celulitis) fueron las más frecuentes (31.5%). Le siguen en frecuencia las complicaciones neurológicas (21%), y las respiratorias (13%) que fueron las más graves, dejando incluso secuelas importantes en el caso de 2 encefalitis. Se hospitalizaron 8 neonatos, tres de ellos para recibir profilaxis con inmunoglobulina por varicela materna y que no desarrollaron la enfermedad. De los otros 5 sólo uno se complicó precisando ingreso en UCIP por neumonía varicelosa. En los casos en los que se extrajo hemocultivo, Streptococcus pyogenes fue el germen aislado con más frecuencia, igual que en los cultivos de lesión cutánea. En cuanto al tratamiento, el 51,7% recibió aciclovir intravenoso y el 57% precisó antibioterapia parenteral. El 8,7% del total de los casos precisó ingreso en UCIP por complicaciones respiratorias o neurológicas. No se produjo ningún fallecimiento. La estancia media hospitalaria fue de 8,6 días.
Conclusiones: Estamos observando un aumento en el número de complicaciones que obligan al ingreso y que en algunos pacientes dejan secuelas graves. Estos motivos apoyan la inclusión de la vacunación sistemática contra la varicela tal y como propone la AEP, la AAP y la mayoría de expertos.
P202 DESCRIPCIÓN DEL IMPACTO EN LA CASUÍSTICA DE MENINGITIS DEL ÁREA DE SALUD 1 DE LA CAM, DE LA VACUNACIÓN FRENTE A N. MENINGITIDIS C A MEDIO PLAZO. 09:35 h
Itziar Marsinyach Ros, Carmen Gutiérrez, Amparo Carreño Beltrán, Rosa Rodríguez, Vicente Climent, Carmen Aritmendi, Gema Arriola Pereda, Kay Boris Brandstrup Azuero, M. Luisa Navarro Gómez, Antonio Gómez
Hospital General Universitario Gregorio Marañón, Madrid.
Antecedentes y objetivo: La enfermedad invasiva por meningococo es un motivo de preocupación a nivel de salud pública a pesar de haber disminuido su incidencia a partir del año 2000. Se plantea, evaluar el impacto de la vacunación universal con vacuna conjugada N. meningitidis C en la casuística de meningitis del área 1 a medio plazo, y valorar las características clínicas y la historia vacunal de los pacientes que desarrollaron enfermedad meningocócica, sospechada o confirmada.
Material y métodos: Se realiza un estudio observacional transversal de revisión de historias clínicas con diagnóstico de meningitis bacteriana entre los años 1998-2003 ambos inclusive. De cada historia clínica se recogieron datos epidemiológicos de vacunación y clínicos-analíticos. Se realizó encuesta telefónica para completar datos de vacunación y factores de riesgo individual.
Resultados: Se realiza análisis descriptivo de los datos. Sobre una muestra de 49 pacientes, 44 tuvieron diagnóstico de meningitis bacteriana y 5 de enfermedad invasiva meningocócica sin meningitis. El 60% tenían entre 3 y 24 meses y el 57,1% eran varones. Se observa un aumento del número de casos en el año 2000 (14) y posteriormente en el año 2003 (9). En un 42.8% de los casos el cultivo de LCR fue estéril. Se confirma una mayor prevalencia de N. meningitidis B respecto a otras etiologías y un aumento del aislamiento de N. meningitidis serogrupo C en el año 2003. Del total, 17 pacientes habían recibido alguna dosis de vacuna conjugada N. meningitidis serogrupo C (34.6%). De ellos 14 (71.4%) desarrollaron enfermedad invasiva probable o confirmada por meningococo. Hubo dos casos de pacientes vacunados con presentación clínica atípica de enfermedad por N. meningitidis C.
Conclusiones: 1) Los datos de incidencia de meningitis bacteriana en este período se asemejan a los comunicados en otras series. 2) Se observa un aumento del número de casos en el año 2003, respecto a los dos previos y un incremento del aislamiento de N. meningitidis serogrupo C en este año. 3) El 71,4% de los pacientes vacunados desarrollaron enfermedad, probable o confirmada por meningococo. 4) El número de LCR estériles es superior al de otras series.
P203 ENCEFALITIS AGUDAS. 09:40 h
Esteban Gómez Sánchez, Ana M. Llorente de la Fuente, Sira Fernández de Miguel, Juan Ignacio Sánchez Díaz, Fernando Mateos Beato, María López Maestro, M. del Carmen García Miranda, M. Victoria Ramos Casado, Sylvia Belda Hofheinz, José Antonio Salinas Sanz
Hospital 12 de Octubre, Madrid.
Introducción: Las encefalitis agudas son procesos inflamatorios que afecta al encéfalo y son desencadenados por un agente infeccioso que puede desencadenarlo de forma directa o a través de una reacción autoinmune. Infrecuentes en la infancia su etiología es difícil de establecer y pueden producir la muerte o dejar secuelas.
Objetivo: Conocer su etiología y el resultado terapéutico en un hospital terciario.
Material y método: Revisión de las HC de los niños con encefalitis aguda infecciosa (EAI) o EMAD atendidos un hospital terciario de Madrid desde enero de 1981 a diciembre de 2002. Se analizaron los estudios microbiológicos realizados, sus resultados y la situación neurológica de los pacientes un año después del ingreso.
Resultados: Desde enero de 1981 hasta diciembre de 2002 hubo 46 casos. Las EAI representaron el 78% y las EMAD el 22%. Se estableció un diagnóstico etiológico en el 41,3% (74% de ellos basado en estudios microbiológicos y el 26% restante en base a clínica de varicela). Se obtuvieron serologías en el 78% de los casos y dieron resultado positivo en un 33%. La detección por PCR en LCR se realizó a partir de 1998 en 11 casos de los 15 posibles (24%) y ofreció un diagnostico en el 18%. El estudio de Ac. Específicos en LCR se realizó en el 11% y fue positivo en el 40%. En todos los casos se procedió al cultivo del LCR sin observarse ningún crecimiento al igual que el cultivo de virus en heces que se realizó solamente en un 19%. Se realizó El cultivo celular de ENF se indicó en el 59% siendo positivo en un 2%. El cultivo de CMV en orina se realizó en un 9% y fue positivo en un 25%. Se practicó una biopsia cerebral (2%) con diagnostico de encefalitis por CMV. Las etiologías establecidas fueron: VHS I, VVZ y VEB 8,7% cada uno, sarampión 6,5%, CMV 4,3%, Strep BHGA y mycoplasma 2,2% cada uno. El 61% de los casos tuvo una evolución favorable quedando libres de secuelas el 67% y un 39% tuvo un mal resultado terapéutico con un 13% de fallecimientos en el global de casos.
Conclusiones: Las encefalitis agudas no son una patología frecuente en la edad pediátrica. El diagnostico etiológico consume muchos recursos y aun así no se alcanza en la mayoría de los casos. Los agentes etiológicos diagnosticados con mayor frecuencia en nuestro estudio pertenecen al grupo herpes y todos ellos son susceptibles de tratamiento etiológico específico. Aún con tratamiento las secuelas son frecuentes y es una patología de alto riesgo vital.
P204 SEPSIS NEONATAL POR STREPTOCOCO AGALACTIAE. REVISIÓN DE CASOS ENTRE 1992-2003. 09:45 h
Àfrica Pertierra Cortada, Rosa Pallàs Ribes, Claudia Fortuny Guasch, Teresa Juncosa Morros, Jordi Pou Fernández
Hospital San Joan de Deu, Barcelona y Universitat de Barcelona, Barcelona.
Objetivos: Revisión epidemiológica de los casos de sepsis neonatal por Streptococo agalactiae.
Material y métodos: Estudio retrospectivo de los casos de sepsis neonatal por Streptococo agalactiae en el Hospital de Sant Joan de Déu de Barcelona entre los años 1992 y 2003.
Introducción: Las infecciones por el Streptococo agalactiae (SGB) constituyen la causa principal de procesos infecciosos graves y mortales en el período neonatal, ocasionando además morbilidad grave, y, con frecuencia, secuelas neurológicas de por vida. En este estudio tratamos de determinar las características clínicas de este proceso, así como los factores de riesgo y el impacto de la profilaxis antibiótica intraparto.
Resultados: Se revisaron 36 pacientes, 17 de ellos fueron sepsis de inicio precoz. La sintomatología más frecuente fue hipertermia (55%), junto con irritabilidad (44%) y rechazo del alimento (36%). Sólo en 7 (19,4%) de los casos la madre era portadora de SGB en el frotis vaginorectal, en el 34.4% de los casos desconocido y en 12 casos no se realizó frotis vaginal/vaginorrectal (8 casos se dieron antes de que se instaurara el cribaje de SGB en gestantes de nuestro centro). En cuanto a la profilaxis antibiótica intraparto, objetivamos que de las 7 pacientes que tenían resultado positivo, 3 fueron tratadas con ampicilina, en 2 casos no consta y en otros 2 casos no se realizó. Los factores de riesgo para la infección por SGB, fueron prematuridad en 9 casos (25%) y RPM > 24h en 4 casos. Tres pacientes, todos ellos con sepsis precoz, fueron éxitus (8,6%); las secuelas a largo plazo fueron, no obstante, más frecuentes en los casos de sepsis tardía con meningitis (problemas de aprendizaje, retraso en el lenguaje, retraso motor, tetraparesia).
Conclusión: Se objetiva un importante descenso de las sepsis precoces neonatales por SGB tras la puesta en marcha del programa de detección de portadoras de SGB (p < 0,05). La prematuridad fue el factor de riesgo asociado a la infección en el 25% de los casos.
NEUROLOGÍA
P205 EFICACIA DIAGNÓSTICA DE UNA NUEVA ESCALA PARA EVALUAR Y PREDECIR LA EVOLUCIÓN DE LA PARÁLISIS FACIAL PERIFÉRICA EN MENORES DE 18 AÑOS. 08:30 h
Lázaro Antonio Ochoa Urdangarain, Olimpo Rodríguez Santos
Hospital Clínico Quirúrgico Provincial Amalia Simoni, Camaguey (Cuba) y Hospital Pediátrico Provincial Eduardo Agramonte Piña, Camaguey (Cuba).
Objetivo: Validar la eficacia diagnóstica de una escala para niños con parálisis facial periférica, cualitativa y cuantitativamente y obtener una función de regresión logística que permita pronosticar a priori su posible evolución.
Antecedentes: En nuestro pais no existe una encuiesta que pueda evaluar satisfactoriamente el pronóstico y evolución del paciente con parálisis facial periférica. Las tareas del médico asistencial son diagnosticar, tratar y emitir un pronóstico. Si reunimos esta información pronóstica podemos definir en que pacientes se debe tomar una conducta conservadora y en cual orientar un tratamiento quirúrgico descompresivo del nervio facial.
Método: Una población de 139 pacientes menores de 18 años con el diagnóstico de parálisis facial periférica a los que se les aplicó la encuesta ya validada por una comisión de expertos constituida por 100 médicos especialistas. Las encuestas fueron sometidas a un análisis de fiabilidad (Programa Statical Package for Social Sciencie) /spss) 10.2 de 1999.
Resultados: En la validación de la encuesta el método ALPHA de COMBRACH arrojó un rango favorable de 0. 8604. Se aplicó una ecuación logística que tiene una efectividad pronóstica de 66,27 que nos orientará en relación al proceder médico ante este paciente. El máximo de efectividad en clasificar correctamente (pronóstico) de la encuesta fue de 70,4% según curva R.O.C (Receiving Operating Charactericts Curve) correspondiente. Los criterios para la valoración de un test (sensibilidad, especificidad, valores predictivos positivos y negativos) ratificaron la utilidad de la encuesta.
Conclusiones: Se ha desarrollado una encuesta útil en más de un 70% para diagnosticar, tratar y definir el futuro de pacientes con parálisis facial periférica.
P206 RETRASO PSICOMOTOR, FALTA DE MEDRO Y MIOCARDIOPATÍA HIPERTRÓFICA SECUNDARIOS A CITOPATÍA MITOCONDRIAL. 08:35 h
Eva Mª Fernández Calderón, Francisco Campo Sampedro, Félix Romero Vivas, Julián Vaquerizo Madrid
Hospital Materno Infantil, Badajoz.
Caso clínico 1: Niña de 14 años diagnosticada de miocardiopatía hipertrófica y Síndrome de Wolf-Parkinson-White tipo b, tras estudio cardilógico de cardiomegalia como hallazgo casual radiológico. Como antecedentes personales de interés destaca retraso ponderoestatural. No antecedentes familiares madre dos abortos. En la exploración se objetiva un retraso psicomotor, gran torpeza motora global, debilidad facial y muscular generalizada, reflejos osteotendinosos vivos, clonus aquíleo bilateral, pares craneales centrados, salvo limitación sutil de la mirada externa del ojo derecho. Láctico, pirúvico, relación lactato/piruvato: elevados en varias determinaciones. Biopsia muscular: Miopatía compatible con citopatia mitocondrial. Determinación de la actividad enzimática en biopsia muscular: déficit de actividad de los complejos I y IV de la cadena respiratoria mitocondrial. Déficit intracelular de carnitina. Estudio de DNA mitocondrial: no mutaciones puntuales ni delecciones compatibles con MELAS o MERRF.
Caso clínico 2: Niño de 2 años que presentó cardiomeglia como hallazgo casual en una radiografía, siendo diagnosticado de miocardiopatía hipertrófica no obstructiva. Antecedentes personales destaca fallo de medro. Clínicamente presentó: hipocinesia, retraso en el inicio de la deambulación, claudición de la marcha, con aumento del tono de miembro inferior derecho, hiperexcitabilidad generalizada, hiperrreflexia con aumento del área reflexojena. Clonus aquíleo agotable, Retraso psicomor. Láctico, pirúvico, relación lactato/piruvato: elevados en varias determinaciones. Biopsia muscular: desproporción congénita del tamaño de las fibras, con deficiencia intracelular de carnitina. Determinación de la actividad enzimática en biopsia muscular: déficit de actividad de los complejos I y IV de la cadena respiratoria mitocondrial con proliferación mitocondrial.
Conclusiones: En ambos casos se objetivó una mejoría en el desarrollo ponderal y psicomotor tras el inicio de tratamiento con cofactores. Consideramos indicado realizar un cribado básico metabólico en procesos pediátricos evolutivos y/o involutivos sin un diagnóstico etiológico o sindrómico especifico, para lo que se podría utilizar la hiperlactacidemia como marcador sensible, aunque no específico.
P207 TROMBOSE VENOSA CEREBRAL EM CRIANÇA, CASO CLÍNICO. 08:40 h
Raquel Guedes, Ana Lopes, António Rui Ribeiro, Fátima Santos, Maria Bom Sucesso
Centro Hospitalar de Vila Nova de Gaia, Portugal.
A trombose venosa cerebral é uma patologia que, apesar de rara na idade pediátrica, tem sido cada vez mais diagnosticada, quer pela maior sensibilização dos clínicos e utilização de técnicas diagnósticas de neuroimagem mais sensíveis, quer pela sobrevivência de crianças com patologias previamente letais que predispõem trombose. Na maioria dos casos envolve os seios cerebrais (septal e transverso), podendo no entanto estender-se às veias cerebrais. A sua etiologia é frequentemente multifactorial, e as variadas e não específicas formas de apresentação tornam a patologia um desafio diagnóstico.Para o seu estabelecimento torna-se, por isso, fundamental um elevado índice de suspeição. A identificação dos factores de risco (adquiridos e congénitos) é fulcral para o seu seguimento. Os autores apresentam um caso clínico de uma criança do sexo feminino de 11 anos de idade, com antecedentes de um internamento por síndrome meníngeo e amígdalite pultácea. É reinternada 3 dias depois por quadro de vómitos incoercíveis, cefaleias e dor abdominal em cólica. Ao exame objectivo encontrava-se apirética sem outras alterações, nomeadamente ao exame neurológico. Após as primeiras 24 horas de internamento verificaram-se alterações do comportamento e flutuações do estado de consciência, sem sintomatologia neurológica focal. Realizou TAC cerebral que revelou imagem de hipodensidade tálamo-capsular direito e suspeita de trombose venosa. Seios peri-nasais sem alterações. A angio-ressonância magnética cerebral confirmou trombose venosa profunda e enfarte venoso talâmico direito. Iniciou heparina em perfusão sendo posteriormente adicionado anticoagulante oral (varfarina). A evolução foi favorável com resolução rápida da sintomatologia. A avaliação complementar de diagnóstico revelou uma heterozigotia para a mutação G20210A da protrombina.
Actualmente em tratamento com warfarina, 8 meses após o diagnóstico, a criança encontra-se assintomática com exame neurológico normal.
Concluindo, perante uma criança com alterações comportamentais e do estado de consciência, é obrigatório pensar no diagnóstico de trombose cerebral, bem como identificar a sua causa pela necessidade de estabelecer um plano terapéutico adequado à situação, evitando complicações a longo prazo.
P208 HIPERGLICINEMIA NO CETÓSICA ATÍPICA Y TRATAMIENTO CON BENZOATO SÓDICO. 08:45 h
M. Pilar González Santiago, Gloria López Lois, María Penín Antón, M. Luisa Murga Sierra, Margarita I. Cebrero García, Carmen Torrijos Román, Magdalena Ugarte Pérez, José Enrique García de Frías
Hospital Príncipe de Asturias, Alcalá de Henares (Madrid), Centro de Diagnóstico de Enfermedades Moleculares, Madrid y Universidad Autónoma, Madrid.
La hiperglicinemia (HG) no cetósica es un error congénito del metabolismo por deficiencia del complejo enzimático glicina-sintasa. Se describen 3 formas clínicas: clásica, transitoria y atípica. La presentación atípica es poco frecuente y tiene dos variantes: forma infantil (con crisis convulsivas y retraso mental a partir de 3-6 meses) y forma tardía (con retraso psicomotor menos intenso, trastorno de comportamiento o síntomas neurodegenerativos). El diagnóstico bioquímico se basa en los niveles elevados de glicina en plasma, orina y LCR con aumento del cociente LCR/plasma (normal < 0,04). El tratamiento con benzoato sódico ha mejorado el control de las crisis y la evolución neurológica en las formas clásicas y atípicas infantiles pero existe poca experiencia sobre la eficacia e indicación de tratamiento en las formas atípicas tardías, sin manifestaciones convulsivas.
Caso clínico: Niña nacida tras embarazo y partos normales; padres sanos y no consanguíneos. Desarrollo psicomotor normal hasta los 2 años aunque presenta episodios de deterioro neurológico desde los 5 meses coincidiendo con procesos febriles. A partir de los 2 años se detecta retraso del lenguaje y problemas de comportamiento y adaptación social. La evaluación psicológica a los 4 años revela un índice general cognitivo de 94 y un desarrollo lingüístico inferior a 3 años.En el estudio neurológico a los 5 años presenta EEG y RNM cerebral normales con elevación de glicina en plasma (411 umol/L), orina (717 mmol/mol creatinina) y LCR (26,6 umol/L) con cociente LCR/plasma 0,065, que permite el diagnóstico de HG no cetósica atípica. Inicialmente se indica restricción proteica moderada y se descartan otras medidas terapéuticas por ausencia de convulsiones. A los 9 años una nueva valoración psicológica indica retraso significativo en habilidades lingüísticas (test de Illinois, Peabody y Concebas) y una valoración conductual con las Escalas de Áreas de Conductas-Problemas para padres y profesores (EACP) detecta problemas de atención sostenida, rendimiento académico y adaptación social. Se comienza tratamienta con benzoato sódico a bajas dosis (250-300 mg/Kg/día) y se repite valoración conductual cada 6 meses observándose mejoría significativa en el comportamiento.
Conclusión: El tratamiento con benzoato sódico a bajas dosis en la HG no cetósica atípica, sin manifestaciones convulsivas, puede mejorar los trastornos de conducta y las dificultades de adaptación escolar y familiar.
P209 MIELITIS TRANSVERSA EN INMUNOCOMPETENTES. 08:50 h
Eider Oñate Vergara, Itziar Sota Busselo, Ramón M. Gaztañaga Expósito, Jesús García Santiago, Agustín Nogues Pérez, Ángeles M. Ruiz Benito
Hospital Donostia, San Sebastián (Guipúzcoa).
Introducción: La mielitis transversa es una enfermedad medular inflamatoria caracterizada por edema y necrosis de uno o varios segmentos. Se manifiesta por disfunción aguda o subaguda motora, sensitiva y autonómica. La incidencia es excepcional.
Observación clínica:

Comentarios: Los hallazgos clínicos y las pruebas complementarias confirman el diagnóstico. No ha podido precisarse ninguna asociación causal. El resultado de la intervención farmacológica ha sido variable. La terapia rehabilitadora debe ser precoz.
P210 CARACTERÍSTICAS EPIDEMIOLÓGICAS Y CLÍNICAS DE LAS CEFALEAS EN LA EDAD PEDIÁTRICA. 08:55 h
Teodoro Durá Travé, M. Eugenia Yoldi Petri, Marta González Villar, Fidel Gallinas Victoriano, Eva Guembero Esarte
Hospital Virgen del Camino, Pamplona (Navarra).
Antecedentes y objetivos: La cefalea infantil es uno de los motivos más frecuentes de la consulta neuropediátrica, representando un problema sanitario de relativa trascendencia. El objetivo del presente trabajo consiste en analizar las características epidemiológicas y clínicas de las cefaleas agudas recurrentes en la edad pediátrica en orden a facilitar su diagnostico diferencial en la práctica clínica diaria.
Métodos: Se han revisado 225 historias clínicas de pacientes con cefaleas agudas recurrentes; recogiéndose características epidemiológicas y clínicas junto a una exploración física y, en su caso, exámenes complementarios. Los criterios diagnósticos aplicados fueron los de la International Headache Society (IHS).
Resultados: El 98,2% de los casos eran cefaleas primarias; migraña (48,9%), cefalea tensional (48,4%) y mixta (0,9%). El 30% de las migrañas tenían aura. La edad de inicio de la migraña era de 8,6 ± 2.9 años, y de la cefalea tensional 9,7 ± 2.5 años (p < 0,05), sin diferencias entre sexos. En la cefalea tensional existía mayor prevalencia (p < 0,05) de sexo femenino, procedencia urbana y rendimiento escolar excelente; y en la migraña existía mayor prevalencia (p < 0,05) de antecedentes familiares. En la migraña el dolor era uni (44,1%) o bilateral (55,9%), pulsátil (77,1%), interrumpía la actividad diaria (65,3%), empeoraba con el ejercicio (68,8%) y se acompañaba de vómitos (71,0%) y fotofobia/sonofobia (67,0%); en la cefalea tensional era bilateral (81,8%), opresivo (85,3%), apenas interrumpía la actividad diaria (11,8%) o empeoraba con ejercicio (25,3%) y ocasionalmente se acompañaba de vómitos (7,3%) o fotofobia/sonofobia (21,8%). Se realizaron EEG y TC craneal en el 21,8% y 39,1% de los pacientes, respectivamente; sin que modificaran el diagnóstico.
Conclusiones: Los tipos más frecuentes de cefalea aguda recurrente en la infancia y adolescencia son la migraña y la cefalea tensional, de inicio preferentemente en la edad escolar; siendo excepcional su relación con patología orgánica grave. Los criterios de la IHS permiten, en gran medida, su diagnóstico diferencial; aunque el control evolutivo sería la prueba de referencia para validar los criterios diagnósticos.
P211 ENFERMEDAD DE LA MIELINA EVANESCENTE. 09:00 h
Mª Lourdes Jiménez Hernández, Idoia Martínez Repáraz, Susana Vidal Piedra, Beatriz Sangrador Martínez, Marta San Román Muñoz, Elena Pérez Gil, Marco Uyaguari Quezada, José Luis Herranz Fernández, Rosa Arteaga Majón
Hospital Universitario Marqués de Valdecilla, Santander (Cantabria).
Introducción: La enfermedad de la mielina evanescente ("vanishing white matter disease") es una nueva entidad clínica descrita en 1993, de herencia autosómica recesiva y etiología desconocida, que suele debutar en la primera infancia.
Caso clínico: Varón primogénito de padres no consanguíneos, sin antecedentes familiares de interés. Embarazo, parto, período neonatal y desarrollo psicomotor normales. A los 23 meses sufre un TCE leve, comenzando al día siguiente con claudicación de la extremidad inferior izquierda y torpeza motora.; tras descartar patología traumática es valorado por neuropediatría. A la exploración destaca: hipotonía muscular de miembros inferiores, reflejos osteotendinosos exaltados, clonus aquíleo izquierdo, claudicación de ambas piernas a la deambulación y genu recurvatum. En la RM se observa leucodistrofia difusa inespecífica intra y supratentorial (ausencia prácticamente total de mielinización). Un mes después, durante una otitis, se produce un rápido deterioro neurológico que lleva a un coma vigil inexplicable precisando traqueostomía y gastrostomía. En la nueva RM cerebral no se aprecian cambios con respecto a la previa. Otras pruebas complementarias: EEG: actividad basal enlentecida y deficientemente configurada; LCR: aumento de glicina; EMG, electroneurografía, potenciales evocados somatosensoriales, visuales y auditivos: normales; estudios metabólicos en suero y orina normales. Tras la recuperación del coma y hasta la actualidad, con cuatro años de edad, permanece en un estado de coma vigil con rigidez generalizada, habiendo padecido en varias ocasiones crisis epilépticas.
Conclusiones: Este cuadro clínico cumple todos los criterios de la enfermedad de evanescencia de la mielina, diagnóstico que debe considerarse en un niño con: 1) desarrollo psicomotor inicial normal, 2) deterioro neurológico progresivo con exacerbaciones espectaculares por trauma craneal leve o infecciones, que pueden llevar a un coma o letargia inexplicables, 3) espasticidad y ataxia progresivas con deterioro mental grave, atrofia óptica y epilepsia, 4) RM con desmielinización rápidamente progresiva de hemisferios cerebrales, 5) aumento de glicina en LCR. Es una enfermedad devastadora que conduce a la muerte en pocos años, ya que sólo existe tratamiento sintomático. La etiología es desconocida, aparentemente genética, habiéndose objetivado mutaciones en el cromosoma 3q27 en algunos pacientes.
P212 UN CASO DE PARÁLISIS FACIAL RECURRENTE POCO FRECUENTE: EL SÍNDROME DE MELKERSSON-ROSENTHAL. 09:05 h
Marta Taida García-Ascaso, Julio Guerrero Fernández, Ana Méndez Echevarría, Ramón Velázquez Fragua
Hospital Materno Infantil La Paz, Madrid.
Introducción: La tríada edema facial y parálisis facial periódicas asociadas a lengua escrotal congénita define una rara entidad que se encuadra dentro del grupo de las parálisis faciales recurrentes: el síndrome de Melkersson-Rosenthal (SMR). Presentamos un caso que cumplía dos de los criterios, suficientes para el diagnóstico de SMR.
Caso clínico: Niño de 10 años de edad que acude al servicio de urgencias por presentar edema hemifacial de predominio labial de varios días de evolución. Los padres relataban dos historias similares que sucedieron, la primera hace tres años y la otra hace uno. Ambas se resolvieron espontáneamente en un corto período de tiempo aunque no pudo llegarse a un diagnóstico concreto. No habían otros antecedentes personales ni familiares relevantes. En la exploración física destacaba edema facial residual que aún podía objetivarse en porción izquierda de labios. De forma casual se descubrió desviación de la comisura bucal hacia el lado contrario cuando se procedía a explorar la cavidad oral; signo de Bell positivo. Resto de la exploración normal.
Discusión: Aunque se presenta de forma esporádica, se han descrito casos familiares de SMR que siguen una herencia de tipo mendeliana autosómica dominante. Puede debutar en la niñez o en la juventud y se manifiesta de forma recurrente como edema facial uni o bilateral de localización predominantemente labial superior e indolente. Puede acompañarse de parálisis facial periférica del mismo lado. La lengua escrotal, no presente en nuestro caso, se encuentra en un tercio de los casos y es de carácter congénito, lo que ayuda, si está presente, al diagnóstico. Nuestro caso constituye una nueva aportación de una entidad excepcional pero de muy fácil diagnóstico y que ha de tenerse en cuenta dentro del diagnóstico diferencial de las parálisis faciales recurrentes.
P213 SINTOMATOLOGÍA NEUROLÓGICA COMO FORMA DE PRESENTACIÓN DE UN TRASTORNO PSIQUIÁTRICO. ¿TRASTORNOS CONVERSIVOS?. 09:10 h
Núria Roig Fort, María Eril, Robert Cilveti, Wilfredo Coroleu Lletget, Marta Azuara Robles, Carlos Rodrigo Gonzalo de Liria
Hospital Germans Trias i Pujol, Badalona (Barcelona).
Objetivo: Se presentan 3 adolescentes con sintomatología neurológica que no responde a ninguna patología orgánica después de la realización de los estudios pertinentes y que se consideran una forma de expresión de los trastornos somatomorfos.
Observaciones clínicas:Caso 1: chica de 16 años con una hemiparesia e hipoestesia facio-braquio-crural derecha de aparición brusca. A la exploración fina hay datos que no concuerdan. Caso 2: chica de 15 años que presenta una paresia y parestesias en extremidad superior derecha. En ambos casos se realizan estudios de imagen que no demuestran ninguna lesión cerebral. Caso 3: chica de 15 años, epiléptica conocida, asintomática en los últimos 3 años. En el último mes reaparecen las crisis comiciales, con una morfología distinta a las habituales, i que no responden a la combinación de 5 anticomiciales de base. Los EEG no muestran cambios significativos respecto los anteriores y el curso de cada episodio comicial es invariable independientemente de las actuaciones médicas que se realicen. En los 3 casos se apuntó hacia un diagnóstico psicopatológico y en la anamnesis se detectaron factores estresantes que podían ser los desencadenantes.
Comentarios: Los trastornos somatomorfos son aquellos que incluyen como característica ciertos síntomas físicos que sugieren una condición médica general y que no se explican completamente por una enfermedad física, ni por los efectos directos de una sustancia ni por ningún otro trastorno mental. Dentro de éstos se incluyen los trastornos conversivos que tienen como rasgos especiales: síntomas que afectan la función motora o sensorial con síntomas psicológicos asociados (factores estresantes desencadenantes), que no son producidos intencionadamente o fingidos y que no se explican por una condición neurológica o médica después de una investigación clínica apropiada. El cuadro completo asocia deterioramiento social, ocupacional o de otras áreas importantes del funcionamiento. Estos trastornos debutan en la adolescencia y si el pediatra es capaz de identificarlos y proporcionar la atención que estos pacientes requieren puede evitar consecuencias disfuncionales importantes. Por otro lado, se debe evitar el error de catalogar un cuadro clínico de somatomorfo sin descartar antes un problema médico.
P214 MOYA-MOYA UN DIAGNÓSTICO POR IMAGEN. 09:15 h
Eider Oñate Vergara, Izaskun Miner Kanflanka, Arantza Vivanco López, Itziar Sota Busselo, Jesús García Santiago, Ramón M. Gaztañaga Expósito, Agustín Nogues Pérez, Ángeles M. Ruiz Benito
Hospital Donostia, San Sebastián (Guipúzcoa).
Introducción: La enfermedad de Moya-Moya es una arteriopatía no inflamatoria del sistema nervioso central de origen desconocido. Se caracteriza por la oclusión de la porción intracraneal de las arterias carótidas internas, con el desarrollo de una amplia red de circulación colateral, dando una imagen en la angiografía cerebral "en bocanada de humo". Su incidencia en Japón se estima en 0,08 casos/100.000 hab/año, siendo desconocida en nuestro medio. Predomina en mujeres (1:1,6) y tiene dos picos de incidencia, a los 4 y 34 años de edad.
Observación clínica:

Comentarios: La etiología se desconoce y se debate si es una enfermedad congénita o adquirida. Se ha descrito asociada a múltiples enfermedades. En los niños la clínica más frecuente es isquémica, siendo más frecuentes los episodios transitorios que los infartos establecidos. Las crisis epilépticas se encuentran en tercer lugar por orden de frecuencia. El tratamiento no está bien establecido. Se han utilizado antiagregantes, anticoagulantes, esteroides y antagonistas del calcio. La tendencia actual, es el empleo de la cirugía para conseguir la revascularización del área isquémica.
P215 CORRELACIONES CLÍNICO-ELECTROENCEFALOGRÁFICAS EN EL SÍNDROME DE ANGELMAN. 09:20 h
M. Ángeles Delgado Rioja, José Sierra Rodríguez, David Mora Navarro, J. Antonio Caballero Romera, Carlos Bueno Ruiz, M. Carmen Menéndez de León, Lucía González Vila
Hospital Juan Ramón Jiménez, Huelva.
Concepto: El síndrome de Angelman es una enfermedad neurológica caracterizada por retardo mental, ausencia del lenguaje, ataxia, dismorfia craneofacial y fenotipo conductual característico (risa inmotivada, apariencia de felicidad y personalidad fácilmente excitable).Con frecuencia presentan convulsiones (80-90%) y alteraciones electroencefalográficas distintivas (ondas lentas hipervoltadas generalizadas, con o sin espigas intercaladas y predominio de localización anterior o posterior). Su etiología es genética, y se han propuesto distintos mecanismos como causantes de éste síndrome, la mayoría (70%) presenta microdelección de la región q11-q13 del cromosoma 15
Pacientes y métodos: Presentamos 4 pacientes (2 varones y 2 mujeres) con diagnóstico de Síndrome de Angelman, confirmado por estudio genético molecular, con edades comprendidas entre los 3 y 10 años. La edad del diagnóstico fue en 2 casos a los 12 meses y a los 1 y 3 años en los otros 2. Evolutivamente, presentaron crisis parciales versivas (1 caso), parciales complejas con automatismos manuales (1 caso), crisis clónicas y mioclónicas generalizadas y status de mal generalizado no convulsivo (2 casos). Todos presentaban un patrón electroencefalográfico típico (ondas delta de amplio voltaje de predominio anterior, entremezcladas con puntas y ondas agudas). El tratamiento combinado con Valproato, asociado a Lamotrigina o Topiramato (3 casos) y con Clonacepam (1 caso), ha logrado en todos un control aceptable de las crisis.
Conclusiones: La asociación de fenotipo físico, conductual y EEG característicos del síndrome de Angelman, conducen a una alta sospecha del mismo, aunque aún no presenten crisis. Esta entidad se incluye dentro de las epilepsias indeterminadas, por presentar tanto crisis parciales como generalizadas, y en nuestros pacientes, status en un 50%. En nuestra experiencia, con la asociación terapéutica de Valproato y Lamotrigina, se ha logrado un buen control de las crisis
P216 ENCEFALOMIELITIS AGUDA DISEMINADA: REVISIÓN CASUÍSTICA DE 5 AÑOS. 09:25 h
Sonia Martínez González, Mª Jesús Martínez González, Ainhoa García Ribes, José M. Prats Viñas
Hospital de Cruces, Barakaldo (Vizcaya).
Introducción: La encefalomielitis aguda diseminada (EMAD) es una enfermedad inflamatoria y desmielinizante del sistema nervioso central, que ocurre más frecuentemente durante la infancia, y que suele estar precedida de un proceso infeccioso o inmunización. El curso suele ser monofásico, aunque pueden aparecer recaídas. La clínica es muy variada, y los estudios complementarios poco específicos, salvo la resonancia magnética cerebral (RNM), que es la prueba de elección.
Pacientes y métodos: Estudio retrospectivo de 9 casos diagnosticados de EMAD en nuestro hospital durante los últimos 5 años (1999-2003), describiendo la forma de presentación, la actitud diagnóstico-terapeútica, y la evolución clínica durante su seguimiento posterior.
Resultados: La edad media de presentación fue de 5 años (rango 1.8-11.8), siendo el 89% varones. El antecedente de proceso infeccioso previo estaba presente en 8 niños. Los síntomas se iniciaron con fiebre en todos los casos excepto en uno. Las manifestaciones neurológicas más frecuentes fueron somnolencia (88,9%), cefalea (66,6%) y convulsiones (44.4%). Dos pacientes presentaron alteraciones en el fondo de ojo. El examen del líquido cefalorraquídeo fue patológico en 8 pacientes, con una pleocitosis media de 71.78 ± 62.48 células, de predominio linfocitario. La RNM mostraba un predominio de afectación de la sustancia blanca subcortical, de distribución multifocal. El curso fue monofásico en 7 niños, presentando los otros 2 una recaída. Se instauró tratamiento con corticoides en 7 casos, corticoides e inmunoglobulinas en una recaída, y tratamiento sintomático en el resto. La evolución fue favorable en la mayoría, con una duración media de 27 ± 19,8 meses de seguimiento.
Comentarios: El diagnóstico de la EMAD es clínico-radiológico, y ante su sospecha se debe practicar una RNM. El tratamiento con corticoides a altas dosis parece ser el más eficaz. Las recaídas pueden aparecer en los primeros 12 meses tras el primer episodio, debiendo hacerse el diagnóstico diferencial con otras enfermedades desmielinizantes como la esclerosis múltiple.
P217 REVISIÓN DE TRASTORNO DE DÉFICIT DE ATENCIÓN CON HIPERACTIVIDAD (TDAH) EN UNA CONSULTA DE NEUROPEDIATRÍA. 09:30 h
Josefina Márquez Fernández, M. Ángeles Carrasco Azcona, Laura Acosta Gordillo, M. del Carmen Medina Gil, M. Ángeles Aguilera Llovet, G. Cruz Guerrero
Hospital Universitario de Valme, Sevilla.
Introducción: El TDAH constituye uno de los procesos más frecuentes que se observan el la consulta de Neuropediatría con un aumento progresivo en los últimos años (actualmente se estima en 5% de las consultas). Se caracteriza por "un patrón persistente de desatención y/o hiperactividad/impulsividad, que es más frecuente y grave que el observado en sujetos con nivel de desarrollo similar". En su etiología están implicados factores tanto biológicos y hereditarios, como psicosociales y estudios recientes señalan la posibilidad de un funcionamiento defectuoso en determinadas regiones cerebrales. Este síndrome puede presentar palología comórbida: trastornos del lenguaje, perceptivo-motores, del aprendizaje o de la conducta. El tratamiento es multidisciplinario: farmacológico, conductual y cognitivo-conductual.
Material y método: Revisión de historias clínicas de 37 niños diagnosticados de TDAH y análisis de las variables: edad, sexo, antecedentes personales y familiares, patología asociada, pruebas complementarias, subtipo y tratamiento.
Resultados: Las edad estaba comprendida entre los 4 y los 15 años. De ellos el 94,5% eran varones. Como antecedentes personales destacamos que un 16,2% presentó convulsiones febriles. En cuanto a la patología asociada, por orden de frecuencia: rasgos dismórficos(35,1%), trastornos del lenguaje(35,1%), trastornos del comportamiento(24,3%), retraso madurativo(18,9%), tics(16,2%), retraso escolar como motivo de consulta(10,8%). Sólo un 13,5% no presentaron trastornos comórbidos.De los 37 niños, en 2 se encontraron alteraciones cromosómicas y otros 2 se diagnosticaron de Sd. De Noonan y Sd. De Rubinstein-Taybi. El 78,3% recibió tratamiento farmacológico. La totalidad de los niños pertenecen a un programa de terapia psicológica de la Unidad de Salud Mental Infantil.
Conclusiones:1) El TDAH constituye un problema socio-sanitario que se observa cada vez con mayor frecuencia en las consultas de pediatría. 2) Es una patología real, que por su variabilidad clínica, repercute en distintos aspectos de la vida del niño y su entorno, y, por tanto, el enfoque terapéutico debe ser combinado: médico, psicológico y psico-pedagógico, lo que posibilita una mejor evolución clínica y una mayor adaptación social.
P218 PARÁLISIS FACIAL CONGÉNITA PERIFÉRICA UNILATERAL. 09:35 h
Inmaculada Sánchez Pérez, Ángel Carrillo Herranz, Juan Manuel Aparicio Meix, Natalia Ramos Sánchez, Manuel Trujillo Peco, Gema de Blas Beorlegui
Hospital Ramón y Cajal, Madrid.
La parálisis facial es una de las mononeuropatías más frecuentes. Su principal etiología es la idiopática (62-93%), seguida de la infecciosa y la postraumática. La congénita, debido a la agenesia de una de las porciones del conducto y nervio facial (como el caso que presentamos) es infrecuente y difícil llegar a un diagnóstico etiológico. Presentamos el caso de una niña de 6 años y 9 meses remitida a nuestro servicio para valoración de parálisis facial periférica derecha. Tras realizar numerosas pruebas complementarias, entre las que se encontraban una TAC y una RMI craneales, en las que no se objetivaron hallazgos patológicos, se optó por un TAC de oídos donde se apreciaba una agenesia de la tercera porción del conducto facial. No se observó afectación de ningún otro par craneal por lo que se descartó un síndrome de Moebius unilateral.
P219 XANTOGRANULOMA JUVENIL CON AFECTACIÓN DE SISTEMA NERVIOSO CENTRAL EN NIÑOS. 09:40 h
Carolina Barreiro Arceiz, M. del Mar Portugués de la Red, Nieves Balado Insunza, Rocío Fernández Martín, Martín Mosteiro Cerviño, Antonio Villadrich Carreira, José Antonio Calviño Castañón, Fernand Vázquez Herrero, Concepción Soler Regal, Jesús Antelo Cortizas
Complejo Hospitalario Xeral-Cíes, Vigo (Pontevedra).
El Xantogranuloma Juvenil es un tipo de Histiocitosis englobado dentro del tipo II o No Langerhans de la clasificación clásica. Su lugar de afectación más frecuente es la piel y los tejidos blandos, aunque puede cursar con alteraciones viscerales. Presentamos el caso de un lactante de 7 meses con Xantogranuloma Juvenil de afectación cerebral, que presenta múltiples lesiones cerebrales hipercaptantes, descubiertas por RM cerebral después de un episodio convulsivo. Se llega al diagnóstico tras un proceso de búsqueda etiológica en que se descartaron causas infecciosas y tumorales de otro tipo, realizándose finalmente una biopsia cerebral. Una vez diagnosticado e iniciado el tratamiento con quimioterapia, aparecen en el niño lesiones cutáneas, que hubieran sido esclarecedoras de haberse presentado antes. Hemos realizado una revisión de la literatura (que exponemos), en la que sólo encontramos 22 casos publicados de Xantogranuloma Juvenil afectando a SNC en niños. Las características especiales de este caso son: se trata de Histiocitosis No Langerhans (menos frecuente); afecta a SNC; de manera múltiple; sin afectación cutánea inicial; se trata de lesiones progresivas a pesar de su benignidad histológica; y su evolución es impredecible, por lo que su tratamiento supone un desafío.
P220 MALFORMAÇÕES DO DESENVOLVIMENTO CORTICAL EM CRIANÇAS COM INFECÇÃO CONGÉNITA POR CITOMEGALOVÍRUS. 09:45 h
Sandra Mesquita, Manuela Santos, Clara Barbot, Inés Carrilho
Hospital de Crianças Maria Pia, Porto (Portugal).
Com o avanço na neuroimagem as malformações do desenvolvimento cortical (MDC), apesar de inicialmente serem consideradas raras, passaram a ser cada vez mais reconhecidas como causa de epilepsia, défices neurológicos e atraso do desenvolvimento. As MDC constituem um espectro de lesões que resultam de agressões de etiologia muito variada ao neocórtex em desenvolvimento. Entre as etiologias possíveis salientamos a infecção congénita pelo citomegalovírus (CMV).
Os autores apresentam 3 crianças com MDC e com o diagnóstico de infecção congénita a CMV. Todos do sexo masculino, com idade actual compreendida entre os 2,5 e 5 anos. As malformações encontradas foram, em duas crianças, polimicrogiria bilateral difusa e noutra uma paquigiria bilateral e esquinzecefalia direita. Clinicamente todas apresentavam atraso do desenvolvimento, duas tinham microcefalia e sinais piramidais, numa criança eram evidentes traços dismórficos além de uma epilepsia. Em dois casos o diagnóstico de infecção congénita por CMV foi feito retrospectivamente (10 meses e aos 2 anos) através da determinação da PCR do CMV no sangue do cartão de Guthrie.
As malformações corticais são uma das possíveis sequelas neurológicas da infecção congénita por CMV. É considerada a infecção congénita mais frequente. As manifestações neurológicas são variadas: microcefalia, tetraparésia, hemiparésia, diplegia, epilepsia, atraso mental, surdez neurossensorial e alterações visuais. Muitas crianças nascem assintomáticas e mais tarde apresentam sequelas da infecção, numa altura em que a determinação das serologias para o CMV não podem estabelecer o diagnóstico de infecção congénita. Os autores alertam para a necessidade de inclusão do CMV nos diagnósticos diferenciais das MDC, dada a sua frequência, salientando o papel do novo método de pesquisa de DNA viral do CMV no cartão de Guthrie que permitirá o diagnóstico retrospectivo da infecção vírica em crianças com alterações neurológicas.
INFECTOLOGÍA
P242 FIEBRE Q: UNA CAUSA POCO FRECUENTE DE ESPLENOMEGALIA. 10:15 h
Ana García González, M. Isabel López Conde, Alba Manjón Herrero, Alicia Cepedano Dans, Esther Vázquez López, Ramón Morales Redondo
Complexo Hospitalario Xeral-Calde, Lugo.
Coxiella burnetii es el agente etiológico de la fiebre Q, una enfermedad que en su forma aguda se manifiesta como fiebre, cefalea, anorexia, tos y en ocasiones esplenomegalia, destacando el componente respiratorio y los síntomas inespecíficos. La variante crónica puede presentarse como hepatitis y/o endocarditis.
Presentamos el caso de una niña de 4 años de edad remitida por su pediatra para estudio por esplenomegalia, llegándose al diagnóstico final de fiebre Q. Procedía de ambiente rural, con animales domésticos en domicilio familiar. Relataba un cuadro de 2-3 días de evolución caracterizado por dolor abdominal con vómitos y diarrea, sin fiebre. Fue diagnosticada de gastroenteritis aguda y tratada con dieta astringente, con buena respuesta. Se realiza exploración física, destacando abdomen blando en el que se palpa bazo a 10 cm. de arcada costal izq., no doloroso ni lobulado, e hígado a 7 cm. de arcada costal dcha. La paciente estaba afebril, con buen estado general y resto de exploración física normal. Se solicitan los siguientes exámenes complementarios: hemograma, frotis sangre periférica, bioquímica (incluido pruebas de función hepática y renal), urocultivo, coprocultivo y aspirado médula ósea, siendo todos ellos normales. Serología para Toxoplasma; Ricketsia; Parvovirus; Citomegalovirus; Brucella; virus de Ebstein-Bar; virus hepatitis (A,B,C); Borrelia; Coxiella burnetii: resultado positivo para esta última (título 1/1280, compatible con infección en actividad). Durante el ingreso presenta fiebre con auscultación cardiopulmonar normal, sin soplos, descartándose endocarditis, la cual es propia de la forma crónica de la enfermedad. Asimismo se descarta hepatitis ya que las pruebas hepáticas fueron normales. Tras diagnosticarse de fiebre Q se inicia tratamiento oral con Eritromicina, con respuesta muy favorable, presentando progresiva disminución de la hepatoesplenomegalia, la cual no se había modificado hasta el momento del tratamiento.
P243 MENINGOENCEFALITIS HERPÉTICA: UN DIANÓSTICO A TENER EN CUENTA. 10:20 h
Raquel Sánchez García
Hospital Universitario Virgen de la Arrixaca, Murcia.
Introducción: La meningoencefalitis herpética es una enfermedad rara, con elevada morbimortalidad, que requiere un diagnóstico y tratamiento precoces, así como un seguimiento posterior adecuado. La forma de inicio es muchas veces atípica, de modo que para su diagnóstico es necesario primero la sospecha clínica y posteriormente la realización de exámenes complementarios apropiados.
Caso clínico: El caso que nos ocupa es el de un varón de 2 años. En sus antecedentes destaca una crisis febril a los 18 meses. Acude a nuestro hospital por cuadro febril y crisis convulsiva intercurrente. En horas posteriores comienza con alteración del comportamiento y episodios repetidos de desconexión del medio de segundos de duración. Durante su ingreso, presenta crisis convulsivas parciales complejas que requieren incluso ingreso en UCIP (estatus convulsivo). Ante la sospecha de meningoencefalitis se instaura tratamiento con aciclovir i.v. pocas horas tras el ingreso manteniéndolo durante 21 días, asi como tratamiento anticomicial con fenitoína y valproato. De forma intercurrente presentó una sepsis por Estafilococo Aureus, secundaria a vía central, que se trato con vancomicina.
Exámenes complementarios más relevantes: 1ª Púnción lumbar (PL): anodina. 2ª PL: 31 leucocitos, 256 hematíes, glucosa y proteínas normales. PCR (reacción en cadena de la polimerasa) para VHS (virus Herpes Simple) en LCR positivo, serología IgG e IgM positivos para VHS. Cultivo de virus y bacterias convencional negativo. RMN: hiperdensidad en T2 en la sustancia blanca de lóbulos temporales mediales. EEG: trastorno irritativo generalizado con predominio a nivel occipital.
Discusión: En un principio se barajaron los diagnósticos de meningoencefalitis, por la fiebre y la sutil alteracion del nivel de conciencia y del comportamiento, debut de epilepsía de puntas occipitales, por los antecedentes de crisis febriles y el trazado del EEG, y crisis febriles atípicas. El diagnóstico de certeza no lo tuvimos hasta tres semanas después del ingreso con el resultado de la PCR en LCR, apoyadas por las imágenes tan sugestivas de la RMN. Al alta el niño estaba asintomático, sin secuelas neurológicas evidentes, pero seguirá revisiones en Neuropediaría, ya que no es infrecuente en esta enfermedad la aparición de déficit psicomotores tras un período de latencia variable.
P244 ADENOFLEMONES CERVICALES POR FUSOBACTERIUM NECROPHORUM ¿SÍNDROME DE LEMIERRE?. 10:25 h
Josefina Díaz Ledo, M. Adela Retana Castán, Antonio Martínez Roig, Nuria López Segura, Verónica Seidel Padilla, Jaime Lozano Blasco
Hospital del Mar, Barcelona y Hospital de la Santa Creu y Sant Pau, Barcelona.
Introducción: Niño de 30 meses que ingresa por presentar tumefacción cervical de 48 horas de evolución y fiebre de 4 días en el contexto de un cuadro catarral de vías altas.
Caso clínico: Sin antecedentes patológicos. A la exploración física presenta hepatomegalia de 4 centímetros sin esplenomegalia, algunos roncus bilaterales, microadenopatías inguinales y adenopatías laterocervicales sin signos inflamatorios, móviles y discretamente dolorosas a la palpación, el resto de la exploración física por aparatos es normal. Exploraciones complementarias: hemocultivo y faringotest negativo, 30000 leucocitos (76% segmentados), PCR 26,5 mg/dl, Anticuerpos Herófilos positivos. Con estos datos se piensa que se trata de una infección vírica en fase inicial y no que se trate de un proceso flemonoso, a pesar de los resultados de los reactantes de fase aguda. A los 4 días del ingreso, con temperaturas superiores a 38,5 ºC, inicia fluctuación de una de las adenopatías con previo aumento del tamaño, se desbridó con cultivo positivo a Fusobacterium Necrophorum, iniciando tratamiento con amoxicilina-clavulánico. A los 3 días, después del descenso parcial de temperatura, reinicia hiperpirexia con fluctuación de un ganglio contralateral que motiva nuevo desbridamiento y se añade clindamicina al tratamiento. Mantiene buen estado general, sin dificultad para la deglución y sin visualización de anomalías en la pared faríngea, con fiebre no superior a 38 ºC. Se realiza TAC cervico-torácico que muestra dos abscesos retrofaríngeos, imágenes nodulares pulmonares bilaterales y ocupación parcial de ambos senos maxilares con niveles hidroaéreos. Ecografía abdominal normal. Se decide no intervenir quirúrgicamente debido al buen estado general y mantenimiento de la apirexia. A las dos semanas el TAC cervical demuestra una disminución importante de los abscesos con casi normalización a los 15 días. Serologías de EBV y CMV negativas. Estudio inmunitario normal.
Conclusiones: ¿Se trata de una infección por Fusobacterium Necrophorum que podría incluirse como una forma incompleta del Síndrome de Lemierre?. En nuestro caso ha existido absceso con foco metastásico, pero sin demostración de bacteriemia ni tromboflebitis y con relativo buen estado general.
P245 ECTIMA GANGRENOSO POR PSEUDOMONA AERUGINOSA EN UN PACIENTE PREVIAMENTE SANO. 10:30 h
Isabel Aguilar Moliner, María Cols Roig, Marta Simó Nebot, Alicia Rodríguez Arráez, Susana Rives Solà, Jordi Pou Fernández
Hospital San Joan de Deu, Barcelona y Universitat de Barcelona, Barcelona.
Introducción: El ectima gangrenoso es una infección cutánea necrotizante que se extiende a la dermis siendo la Pseudomona aeruginosa el principal germen implicado. La localización más frecuente es la región perineal por vía hematógena. Aunque no es frecuente que se asocie a septicemia, cuando es así la mortalidad se eleva a más del 50%. El diagnóstico se realiza a través del cultivo de una muestra de la lesión y/o del hemocultivo. El tratamiento de elección se basa en la asociación de dos antibióticos antipseudomónicos por vía parenteral. Afecta principalmente a immunocomprometidos siendo excepcional en pacientes previamente sanos.
Caso clínico: Niña de 18 meses que presenta fiebre de 5 días de evolución, cuadro catarral y deposiciones dispépticas. Como antecedentes de interés destacan dos episodios de infección de orina sin uropatía de base. En la exploración física al ingreso se observa una discreta celulitis en labios genitales mayor y menor izquierdos, siendo el resto de la exploración normal. En la analítica sanguínea se detecta una neutropenia intensa y PCR de 155,5 mg/L. Ingresa con cobertura antibiótica empírica por síndrome febril en paciente neutropénico (ceftazidima y amikacina iv). Se realizan hemocultivos, radiografía de tórax, sedimento de orina y urocultivo que son normales. Se realiza despistaje de enfermedades immunosupresoras: poblaciones linfoides e immunoglobulinas normales, serologías a VHB, VIH, CMV, Parvovirus B19, Toxoplasma, Herpes negativas, immunidad adquirida a Rubeola y a VEB con Paul-Bunnell negativo. Al día siguiente del ingreso aparece una úlcera en el labio menor genital con aumento de la celulitis en el labio mayor. Se recoge un frotis del exudado para cultivo creciendo P.aeruginosa sensible a la antibioterapia empleada. La paciente presenta una buena evolución clínica con desaparición de la fiebre al tercer día y mejoría progresiva de la úlcera genital. Se da de alta tras 15 días de tratamiento, con normalización del hemograma y la lesión genital en proceso de cicatrización.
Comentarios:1) Ante una infección por P.aeruginosa en un niño previamente sano se deberían descartar posibles estados de immunosupresión. 2) Ante la sospecha de ectima gangrenoso, el diagnóstico y el tratamiento deben ser precoces ya que la bacteriemia por P. aeruginosa conlleva una alta mortalidad en niños (15-28%).
P246 VARICELA Y HERPES ZOSTER SIMULTÁNEOS EN NIÑA INMUNOCOMPETENTE. 10:35 h
Idoia Martínez Repáraz, Beatriz Sangrador Martínez, Mª Lourdes Jiménez Hernández, Susana Vidal Piedra, Mª José Lozano de la Torre, Ángel Pérez Puente, Vicente Madrigal Díez, Lucía Díaz de Entresotos Villazán, Mercedes Sánchez Rodríguez
Hospital Universitario Marqués de Valdecilla, Santander (Cantabria).
Introducción: La infección por el virus varicela-zóster (VVZ) se manifiesta clínicamente de dos formas: A) Varicela, forma generalizada que acompaña a la primoinfección con viremia. B) Herpes zóster, forma localizada producida por reactivación ocasional de virus latentes en los ganglios nerviosos tras la primoinfección. Presentamos el caso de una niña que presentó simultáneamente ambos cuadros clínicos.
Caso clínico: Niña de 9 años sin antecedentes de varicela previa, cuyo padre y hermano la padecieron cuando ella tenía 3 semanas de edad, que comenzó, ocho días antes del ingreso, con eritema pruriginoso en región cervical posterior derecha extendiéndose en los días sucesivos hacia la parte anterior del cuello y cara, adquiriendo carácter vesicular confluente. En las últimas 24 horas brotaron lesiones pápulo-vesiculosas diseminadas en tronco y raíz de miembros.
Exploración física: Niña con normal desarrollo pondoestatural. Buen estado general. Eritema exudativo micropapuloso confluente que afectaba al territorio de segunda y tercera rama del trigémino derecho y en algunas zonas lesiones vesículo-pustulosas que evolucionaron hacia costras no sobrepasando la línea media. En tronco, raíz de los miembros y cuero cabelludo se apreciaba una erupción pápulo-vesiculosa diseminada con elementos aislados y sin tendencia a confluir, en diferentes estadios evolutivos, característica de varicela.
Exámenes complementarios: Serología VVZ: IgG e IgM positivas. El estudio inmunológico resultó normal.
Evolución y tratamiento: Instaurado tratamiento con aciclovir, las lesiones evolucionaron favorablemente.
Conclusión: Se ha descrito algún caso de padecimiento simultáneo de varicela y herpes zóster en pacientes inmunodeprimidos. En nuestro caso, tratándose de niña inmunocompetente, pensamos que padeció una infección asintomática en el período neonatal (contagiada por sus familiares) cuando estaba protegida parcialmente por los anticuerpos maternos. Probablemente estas circunstancias condicionaron que la respuesta inmune fuera insuficiente. Esto hizo que en la reactivación de los virus latentes se produjera una enfermedad local, el herpes zóster, con lesiones muy intensas y una viremia que originó un cuadro de varicela típica.
P247 OSTEOMIELITIS HEMATÓGENA AGUDA. ASPECTOS EPIDEMIOLÓGICOS, CLÍNICOS, RADIOLÓGICOS Y TERAPÉUTICOS. 10:40 h
Víctor Manuel Navas López, David Canalejo González, Juan David González Rodríguez, Inmaculada Guillén Rodríguez, Estrella Peromingo Matute, Aurelio Cayuela Domínguez, M. Soledad Camacho Lovillo, Cristina Montero Valladares, Juan Antonio León Leal, Juan Navarro González
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivo: La osteomielitis hematógena aguda es la forma mas frecuente de osteomielitis en la infancia. No está exenta de complicaciones ni secuelas. Requiere diagnóstico precoz y tratamiento enérgico. Determinar frecuencia, características clínicas y radiológicas, sensibilidad de las distintas pruebas complementarias.
Material y método: Estudio descriptivo transversal del período 1994-2003 que incluye a todos los pacientes menores de 15 años diagnosticados de osteomielitis hematógena aguda en el servicio de Infectología del Hospital Infantil Virgen del Rocío de Sevilla.
Resultados: La muestra objeto del estudio está formada por 38 pacientes, con una edad mediana de 6,64 años (rango intercuartílico 1,31-10,44); de los cuales 21 varones (52,8%) y 17 mujeres (47,2%). Los meses con mayor incidencia fueron los de primavera y verano. Se recogió historia previa de traumatismo en el 30,6% de los pacientes, con un tiempo medio entre el trauma y la aparición de la sintomatología de osteomielitis de 3 días. Los huesos mas comúnmente afectados fueron tibia (26,8%), fémur (19,5%) y calcáneo (9,8%), localizándose el foco séptico con más frecuencia en la metáfisis (34,1%) seguido de la epífisis (27,3%). En 2 neonatos había afectación del hueso parietal. En tres casos la afectación ósea fue múltiple. En 8 pacientes (21,1%), junto a la osteomielitis se constató afectación articular. Las manifestaciones clínicas más frecuentes fueron el dolor (86,8%) y la impotencia funcional (73,7%). La PCR se cuantificó en el 44,7% de los pacientes, en todos ellos se encontraba elevada. Los hallazgos radiológicos más frecuentes fueron la osteolisis y la disminución de la densidad ósea. La gammagrafía ósea y la RNM fueron patológicas en todos los pacientes, la TC en más del 90%. En el 60% de los pacientes se aisló S. Aureus meticilin sensible. La duración mediana del tratamiento antibiótico fue de 27,50 días (rango 23-34). La evolución fue favorable en el 80% de los pacientes, 13 pacientes necesitaron tratamiento quirúrgico.
Comentarios: Es obligado un diagnostico precoz y un tratamiento enérgico a fin de evitar las secuelas en la edad adulta. El tratamiento antibiótico empírico debe instaurarse siguiendo criterios epidemiológicos y microbiológicos.
P248 SÍNDROME DE LA ESCALDADURA ESTAFILOCÓCICA. IMPORTANCIA DEL DIAGNÓSTICO POR LA CLÍNICA. 10:45 h
Susana Vidal Piedra, Beatriz Sangrador Martínez, Idoia Martínez Repáraz, Mª Lourdes Jiménez Hernández, Carmen Madrigal Díez, Marta San Román Muñoz, Marco Uyaguari Quezada, Mª José Lozano de la Torre, Ángel Pérez Puente, Vicente Madrigal Díez
Hospital Universitario Marqués de Valdecilla, Santander (Cantabria).
Introducción: Se denomina síndrome de la piel escaldada a un grupo de trastornos ampollosos de la piel provocados por toxinas exfoliativas producidas por determinadas cepas de Staphylococus aureus. Es un síndrome poco frecuente que afecta preferentemente a recién nacidos y menores de 5 años. Su diagnóstico es fundamentalmente clínico. Los cultivos de las lesiones cutáneas suelen ser estériles ya que la toxina se libera desde el foco infeccioso y se disemina por vía hematógena.
Caso clínico: Niño de 5 años con cuadro de 36 horas de evolución con febrícula y exantema, inicialmente eritematoso y papuloso, en región cervical, axilar e inguinal, que fue interpretado como urticarial. Posteriormente apareció dolor y se apreció componente exudativo en algunas lesiones, junto con edema palpebral y fisuración labial. En las horas siguientes se inició despegamiento de epidermis. A nivel de la cola de la ceja derecha presentaba una pequeña erosión que parecía sobreinfectada. Pruebas complementarias: Hemograma con discreta leucocitosis. Cultivo de frotis faríngeo: S. aureus. Cultivo del exudado de las lesiones: negativo. Se realizó tratamiento con cloxacilina intravenosa y curas locales con mupirocina, siendo la evolución clínica favorable.
Conclusiones: Conviene tener en cuenta que en las fases iniciales el síndrome de la escaldadura estafilocócica puede pasar desapercibido por su parecido con otras afecciones cutáneas. La instauración precoz del tratamiento acorta la evolución y mejora el pronóstico de la enfermedad, de ahí la importancia de evocar su existencia ante los datos de una cuidadosa anamnesis y de la exploración física, más que por los proporcionados por pruebas complementarias.
P249 ADENOPATÍAS SUBMANDIBULARES Y MYCOBACTERIUM AVIUM COMPLEX EN PACIENTE INMUNOCOMPETENTE. 10:50 h
Olaia Sardón Prado, Nere Arostegi Kareaga, Carmen García Pardos, Ángeles M. Ruiz Benito, Andrea Bordoy Riera, Francisco Javier Mintegui Aramburu, Esperanza Pérez Ruiz
Hospital Donostia, San Sebastián (Guipúzcoa).
Introducción: La infección por micobacterias atípicas en inmunocompetentes está presentando un incremento en los últimos diez años (F de Juan Martin et all. An Ped 2002). Están incluidas en la etiología de las adenitis cervicales.
Caso clínico: Niña de 2 años de edad que presenta desde hace 7 días, dos adenopatías submaxilares izquierdas de 1,5 x 2 cm de diámetro, de aspecto eritematoso y consistencia leñosa, sin modificación tras tratamiento con amoxicilina-clavulánico vía oral. No enfermedades previas de interés. Inmunización correcta (excepto BCG).
Exámenes complementarios primer nivel (Cruz 2003): hemograma, VSG, transaminasas, serología, frotis faríngeo y radiografía de tórax: normal. Mantoux 13 mm. Segundo nivel: LDH, ecografía cervical y baciloscopia de contenido gástrico: negativo. Examen de ganglio por punción aspiración con aguja fina (PAAF): Bacilos Ácido Alcohol Resistentes (BAAR). Inicialmente, se instauró tratamiento médico dirigido a Mycobacterium tuberculosis, hasta la identificación definitiva de Mycobacterium avium complex. Ante la imposibilidad de exéresis quirúrgica completa debido al aumento de tamaño y la fistulización espontánea; se instauró tratamiento médico específico con claritromicina y rifampicina, con mejoría clínica.
Comentarios: 1) Importancia de incluir las micobacterias atípicas en el diagnóstico diferencial de adenopatías cervicales de causa infecciosa en niños inmunocompetentes. 2) Valorar la realización de PAAF como método diagnóstico ante adenopatías de crecimiento rápido, no disminución de éstas tras 2-4 semanas de tratamiento antibiótico, ó localización supraclavicular. 3) El tratamiento de elección de adenopatías por micobacterias atípicas, es la exéresis quirúrgica completa. El tratamiento médico con claritromicina y rifampicina estaría indicado, cuando ésta no es posible.
P250 EL PLAN DE ELIMINACIÓN DEL SARAMPIÓN EN CANARIAS. EVALUACIÓN DE SUS RESULTADOS. 2001-2003. 10:55 h
Amós José García Rojas, Pilar García Castellano, María Dolores Trujillo Herrera, José Solís Romero, Petra Matute Cruz, Natividad Abadía Abascal, Lucas González Santacruz, María del Carmen Pérez González
Dirección General de Salud Pública, Las Palmas y Hospital Universitario Dr. Negrín, Las Palmas de Gran Canaria (Las Palmas).
Antecedentes: El Plan de Eliminación del Sarampión en Canarias se puso en marcha en Enero de 2001, siguiendo las recomendaciones establecidas por OMS, lo determinado en el Plan de Acción para eliminar el Sarampión en España y a los objetivos del Plan de Salud de Canarias. Se caracteriza por una triple vertiente de actuaciones, A) Póliticas de vacunación, B) Vigilancia Epidemiológica y C) Laboratorio. Se describen los resultados obtenidos en el trieno 2001-2003
Métodos: Se analizaron los resultados de cobertura con Triple Vírica obtenidos durante esos años, utilizando como denominador al conjunto de niños/as con edades comprendidas entre los 1 y 2 años, y como denominador al conjunto de niños/as de esas mismas edades vacunadas con Triple Vírica. Así mismo, se revisaron los casos de sarampión notificados a la Red Canaria de Vigilancia Epidemiológica, desde las 0 horas del 1 de Enero de 2001 a las 24 horas del 31 de Diciembre de 2003, obtenidos de forma individualizada según ficha epidemiológica establecida en el Plan.
Resultados: Las coberturas obtenidas con Triple Vírica durante los tres años estudiados, han sido siempre superiores al 90%. Por otro lado, se notificaron 36 casos sospechosos de sarampión, de los cuales 34 se clasificaron por laboratorio (31 descartados y 3 confirmados). Los 3 casos confirmados eran importados.
Conclusiones: Las altas coberturas conseguidas para la vacuna Triple Vírica, junto al hecho de que desde la puesta en marcha del Plan no se ha declarado ningún caso autóctono de esta enfermedad en Canarias, nos indican lo adecuado del funcionamiento del Plan.
P251 NÓDULO TIROIDEO SECUNDARIO A INFECCIÓN POR ASPERGILLUS FUMIGATUS. 11:00 h
Marta Taida García-Ascaso, Julio Guerrero Fernández, Marta Melgosa Hijosa, Carlota Fernández, Carmen Meseguer, Laura Espinosa, Ángel Alonso, Antonia Peña, Mercedes Navarro
Hospital Materno Infantil La Paz, Madrid.
El tiroides se puede afectar por procesos infecciosos de tipo viral, bacteriano o fúngico. En algunas ocasiones la afectación de la glándula da lugar a clínica tiroidea por invasión y destrucción de la misma. Presentamos un caso de tirotoxicosis asociado a infección fúngica localizada por Aspergillus fumigatus.
Caso clínico: Niña de 10 años, proveniente de Mauritania, diagnosticada de Lupus Eritematoso Sistémico con nefropatía en fase terminal ingresado en nuestro Servicio para valoración y tratamiento con hemodiálisis. En la exploración llama la atención un latido hiperdinámico con tensión arterial elevada y soplo sistólico, así como un nódulo tiroideo palpable de agrandamiento progresivo. Determinaciones analíticas: TSH = 0,01 UI/mL (VN: 0,25-6,15); T4L = 4,78 ng/dL (VN: 0,70-1,64); T3L = 1,10 ng/dL; TSI = 1; Tiroglobulina = 523; anticuerpos antitiroideos < 60 (negativos). Se realiza ecografía tiroidea encontrándose un lóbulo derecho de gran tamaño próximo a la canalización de una vía central, haciéndonos pensar en un primer momento en un posible traumatismo quirúrgico de la misma. A los 29 días del ingreso, ante la persistencia del nódulo y de la alteración hormonal, se realiza gammagrafía con tecnecio hallándose un nódulo frío de grandes dimensiones en el lóbulo tiroideo derecho. Posteriormente se le realiza una punción-aspiración con aguja fina, observándose en la muestra abundantes hifas sugerentes de Aspergillus fumigatus y confirmándose posteriormente con el cultivo de la misma. No se observan focos de afectación por hongos a otros niveles. La clínica de tirotoxicosis se controla durante el ingreso con atenolol, no requiriendo tratamiento antitiroideo específico. Durante la estancia hospitalaria se van normalizando progresivamente los niveles de hormonas tiroideas, así como se observa disminución progresiva del tamaño del nódulo y desaparición de la clínica. A pesar de la resolución espontánea y la falta de afectación sistémica de origen fúngico dada la agresividad del germen y la situación de inmunosupresión, se decide iniciar tratamiento de la infección con itraconazol a 4 mg/kg/día por vía oral.
Conclusión: Por lo revisado en la literatura, es excepcional el caso de infección tiroidea de origen fúngico, siendo todavía más singular la falta de repercusión sistémica en una infección por Aspergillus fumigatus en una paciente de tales características.
P252 COINFECCIÓN POR BORDETELLA PERTUSSIS Y VRS EN UN HOSPITAL TERCIARIO: REVISIÓN DE 10 AÑOS. 11:05 h
Ana Gómez Zamora, Marta García Fernández de Villalta, Fernando Baquero Artigao, Beatriz Larrú Martínez, M. Isabel de José Gómez, Clementina Borque Andrés, María Jesús García de Miguel, Francisco Martínez Cortés, Fernando del Castillo Martín
Hospital Materno Infantil La Paz, Madrid.
Objetivos: Estudiar los casos de coinfección por Bordetella pertussis y virus respiratorio sincitial, analizando su repercusión sobre la evolución clínica y la estancia hospitalaria.
Métodos: Estudio retrospectivo de los niños con tos ferina confirmada por cultivo nasofaringeo a los que se había realizado de forma simultánea la determinación del virus respiratorio sincitial en moco nasal, durante un período de diez años (1994-2003). Se ha realizado un estudio estadístico comparativo de las características clínicas, complicaciones y estancia hospitalaria del grupo con coinfección frente al grupo con VRS negativo.
Resultados: Se identificaron 78 pacientes con cultivo positivo para Bordetella pertussis. En 31 casos (39%) se había realizado la determinación de VRS en moco nasal de forma simultánea. Se obtuvieron 6 casos de coinfección Bordetella-VRS (20%), todos ellos entre los meses de enero a mayo. En cuanto a las características clínicas y analíticas de ambos grupos, no se encontraron diferencias estadísticamente significativas en cuanto a la presencia de cianosis, vómitos, gallo, apnea, linfocitosis, auscultación y radiografía patológica. Únicamente se constató una mayor frecuencia de dificultad respiratoria (p < 0,002) en el grupo con coinfección y una tendencia no significativa a una mayor necesidad de oxigeno (5,5 días frente a 2 días), así como a una mayor estancia hospitalaria (15,7 frente a 10,7 días). Un 10% de los pacientes precisaron el ingreso en la unidad de cuidados intensivos pediátricos, no evindenciándose diferencias estadísticamente significativas entre ambos grupos.
Conclusiones: La frecuencia de coinfección entre Bordetella y VRS en nuestro medio es elevada. Aunque la coinfección parece agravar el curso de la tos ferina, no se han obtenido diferencias significativas posiblemente debido al pequeño tamaño muestral.
P253 OSTEOMIELITIS POR STAPHYLOCOCCUS AUREUS RESISTENTE A LA OXACILINA ADQUIRIDO EN LA COMUNIDAD. 11:10 h
Cristina Ruiz Serrano, Mª Verísima Barajas Sánchez, Miriam Blanco Rodríguez, Ruth González Crisóstomo, José Manuel Sánchez Granados, M. Elena Zamora Flores, Mercedes Bernacer Borja
Fundación Jiménez Díaz, Madrid.
Introducción: La incidencia de infección por Staphylococcus aureus resistente a oxacilina (SAOR) es actualmente excepcional en nuestro medio, a pesar de que cada vez son más numerosos los casos comunicados de infecciones adquiridas en la comunidad por esta causa.
Caso clínico: Se trata de un escolar de diez años de edad diagnosticado de epifisiolisis traumática del tercio distal del radio derecho, sin solución de continuidad en la piel. A los tres días consulta por presentar febrícula, dolor y tumefacción local. Tras la retirada de la férula de inmovilización se objetivan signos inflamatorios locales, por lo que se decide su ingreso y administración de antibioterapia endovenosa con amoxicilina-clavulánico, previa recogida de hemocultivo. A las 48 horas se informa del crecimiento de SAOR, motivo por el que se sustituye el tratamiento por vancomicina. Se realiza gammagrafía con Tc-99 y leucocitos marcados sin evidenciar captación ósea. Al séptimo día de tratamiento presenta una masa fluctuante en el tercio distal del radio, objetivándose ecográficamente la presencia de un absceso subcutáneo. Se drena quirúrgicamente, con confirmación microbiológica posterior de la etiología estafilocócica y tras 14 días de tratamiento endovenoso, el paciente es dado de alta. A las tres semanas, reingresa por presentar dolor y tumefacción local, sin otros signos inflamatorios. Se evidencia una lesión osteolítica en el tercio distal del radio mediante radiología. realizándose limpieza y desbridamiento quirúrgico, con crecimiento de SAOR en la muestra ósea. El paciente es tratado con Vancomicina y Rifampicina intravenosas durante tres semanas y posteriormente Clindamicina oral cuatro semanas.
Comentarios:1) La enfermedad invasora por SAOR adquirida en la comunidad es excepcional en nuestro medio. Actualmente hay un aumento en la incidencia de infecciones por este microorganismo. 2) En nuestro caso el paciente fue inicialmente infratratado durante el primer ingreso al no tener evidencia de afectación ósea, lo que justificó la mala evolución posterior.
P254 A PROPÓSITO DE UN CASO DE ESCLEREDEMA ADULTORUN DE BUSHKE. 11:15 h
Carmen Fresneda Machado, José M. López Gómez, Vicente J. Ramírez Piedrabuena, M. Teresa Moya Díaz-Pintado, Mª Sierra Antona García, Arturo Sánchez Enfedaque
Hospital Virgen de Altagracia de Manzanares, Ciudad Real.
Se presenta el caso de una niña de 2 años y medio sin antecedentes personales ni familiares de interés que acude a urgencias por un cuadro de fiebre de 38ºC de 4 días de evolución secundario a foco O.R.L. con induración cutánea en cara y cuello de 3 días de evolución.
Exploración: BEG. Buen color de piel y mucosas, bien nutrida e hidratada, TA: 110/60. Induración cutánea llamativa sin fovea en cara, cuello, tórax y miembros superiores con limitación de la abertura ocular y bucal. Auscultación cardiopulmonar, exploraciones abdominales y neurológicas normales. Supuración purulenta en oído izquierdo con amígdalas hipertróficas e hiperémicas.
Pruebas complementarias al ingreso:a) Hemograma y bioquímica: normales, PCR: 1.7 mg/dl, Paul-Bunnell: negativo, b) Electrocardiograma: normal, c) Rx de tórax: normal.
Evolución y tratamiento: Ante la sospecha clínica de escleredema adultorun de Bushke se instaura tratamiento con corticoides a 2 mg/Kg/día así como cefotaxima y ibuprofeno como tratamiento del proceso infeccioso.
Durante su ingreso en nuestro hospital la niña presenta progresión rápida de la induración cutánea en todo hemicuerpo superior. Se traslada a otro Hospital para realizar Ecocardiografía y confirmación diagnástica. El estudio cardiológico ha sido normal. Se ha realizado biopsia cutánea que es compatible con dicho cuadro. Actualmente, 4 meses tras el diagnóstico, continúa tratamiento con corticoides orales, persistiendo la induración cutánea, no ha presentado complicaciones sistémicas. Realiza revisiones periódicas en Cardiología, Dermatología y Reumatología pediátricas. El interés de este caso radica en su escasa frecuencia. El cadro cutáneo es muy característico y sobre todo su rápida progresión inicial. La causa más frecuente es como complicación de una infección de vías respiratorias superiores aunque hay que descartar otras causa graves. Suele resolverse en semanas o meses pero requiere seguimientos estrechos dados sus graves complicaciones sistémicas.
P255 ENDOCARDITIS INFECCIOSA A CÁNDIDA PARAPSILOSIS CON BUENA EVOLUCIÓN BAJO TRATAMIENTO MÉDICO. 11:20 h
Antonio Gómez Calzado, J. Moya Angelec, Antonio González-Meneses López, R. Moreno de Alba, Fidel Plaza García, Francisco Chaves Pecero, José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Objetivo: Presentación de un caso de endocarditis infecciosa nosocomial a Cándida parapsilosis que ha seguido una buena evolución bajo tratamiento médico.
Material y métodos: Niño de 8 años de edad, afecto de fístula enterocutánea postoperatoria debida a dehiscencia parcial de sutura de anastomosis intestinal, aparecida durante el postoperatorio de una necrosis intestinal secundaria a brida sobre divertículo de Meckel. Tras 7 días bajo tratamiento conservador con alimentación parenteral, por vía venosa periférica, y somatostatina, apareció un cuadro febril de larga duración, acompañado de funguemia persistente, revelada por hemocultivos positivos a Cándida parapsilosis desde el primer día de fiebre, sensible a Fluconazol y a Anfotericina B. Por indicación del Servicio de Microbiología, recibió tratamiento con Fluconazol, que se mantuvo durante un total de 12 días, tras lo cual se cambió a Anfotericina B complejo lipídico debido a persistir la fiebre. Tras 13 días de fiebre, se practicó ecocardiografía transtorácica, encontrándose un conglomerado ecogénico, brillante, en velo anterior tricuspídeo, de 11,4 x 7,1 mm de diámetro. Este hallazgo ecocardiográfico y la funguemia persistente antes referida, cumplen los 2 criterios mayores de los de la Universidad de Duke, que definen el diagnóstico de endocarditis infecciosa por criterios clínicos.
Resultados: La fiebre desapareció a los 19 días de evolución, con hemocultivos negativos. El tamaño de la vegetación endocárdica, por ecocardiografía, disminuyó progresivamente (tras 52 días afebril, media 6 mm de diámetro y estaba calcificada).
Conclusiones: Aunque es difícil saber la incidencia de la endocarditis infecciosa en niños, parece que ha aumentado, especialmente por un mayor número de casos nosocomiales, con o sin anomalías cardíacas subyacentes, ligados al mayor número de pacientes con catéter venoso central o alimentación parenteral. Han aumentado los casos de endocarditis fúngica. Actualmente, la Ecocardiografía permite el diagnóstico precoz de la endocarditis. Hay varios casos descritos de endocarditis fúngica con buena evolución bajo tratamiento médico (como ocurre en el caso que presentamos), sin necesidad de tratamiento quirúrgico.
P256 VARICELA: EPIDEMIOLOGÍA Y COMPLICACIONES. 11:25 h
Concepción Hidalgo Figueroa, María Jesús Balboa Vega, Isabel M. Palma Fuentes, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio, José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Objetivos: Conocer la incidencia de varicela en nuestro medio y determinar las causas que motivan su hospitalización.
Material y métodos: Estudio retrospectivo de los niños que acudieron al Servicio de Urgencias de nuestro Hospital con varicela entre enero1990 y diciembre 2003. Se revisan datos sobre la edad, sexo y fecha de consulta. En los niños ingresados se estudian, además, antecedentes personales, complicaciones desarrolladas, tratamiento y días de estancia hospitalaria.
Resultados: En el período estudiado 1317 niños consultaron por varicela en nuestra Urgencia. La mayor incidencia ocurrió en el año 1999 y fueron los meses de marzo a julio donde ésta se presentó con más frecuencia (71,2%). Del total de consultas, ingresaron 153 niños (11,6%), con máxima incidencia en el grupo de edad de 1 a 4 años (48,4%). Los motivos más frecuentes de ingreso se pueden dividir en tres grupos: 1) Afectación importante del estado general con lesiones generalizadas e hipertermia (78%). 2) Desarrollo de complicaciones propias de la varicela: sobreinfección de la piel y tejido celular subcutáneo, convulsión febril, blefaroconjuntivitis, neumonía, encefalitis, ataxia cerebelosa, glomerulonefritis, púrpura postinfecciosa, artritis y síndrome de coagulación intravascular diseminada (50%). 3) Niños con factores de riesgo para desarrollar una varicela grave: niños menores de un año, síndrome nefrótico con altas dosis de corticoides, infección VIH, artritis crónica juvenil en tratamiento con salicilatos y broncopatía aguda disneizante de repetición (23,5%). La estancia media hospitalaria fue de 7 días. La evolución fue favorable en todos los casos excepto una niña inmunodeprimida que falleció por una neumonía. No se encontraron secuelas secundarias a la enfermedad.
Conclusiones: A pesar de su benignidad, un número significativo de niños requirió hospitalización. El motivo más frecuente de ingreso fue la afectación del estado general. La mayoría eran niños sanos previamente. La complicación más frecuente fue la sobreinfección cutánea. El tiempo de estancia hospitalaria es similar al de otras series revisadas.La vacunación universal supondría una disminución de la morbimortalidad así como una reducción del coste social y sanitario, que en atención primaria en España supone 96,2euros entre gastos directos e indirectos en cada caso.
URGENCIAS
P258 ACCIDENTES DE TRÁFICO EN NIÑOS: IMPACTO EN UN REGISTRO DE TRAUMA PEDIÁTRICO. 10:20 h
Iván Somoza Argibay, Alberto Sánchez Abuín, Jorge Liras Muñoz, Roberto Méndez Gallart, Manuel Gómez Tellado, Ernesto Pais Piñeiro, José Ríos Tallón, Diego Vela Nieto
Hospital Juan Canalejo, A Coruña.
Introducción: Los traumatismos continúan siendo la causa más frecuente de mortalidad infantil en la mayoría de los países y en nuestra comunidad; entre ellos los derivados de los accidentes de tráfico destacan por su frecuencia y mayor mortalidad. El 50% de esas muertes por accidente son prevenibles. Constituyen un grave problema de salud pública que precisa soluciones urgentes.
Material y métodos: Entre Abril de 2001 y Enero de 2003 se trataron en nuestro Hospital 11.128 niños de menos de 14 años por traumatismos agudos. De ellos 637 pacientes fueron ingresados e incluidos en nuestra base de datos. 178 fueron traumatismos derivados de accidentes de circulación y fueron sometidos a estudio. Todos los pacientes registrados tras su ingreso en el Servicio de Urgencias, fueron clasificados según su gravedad siguiendo el Índice de Trauma Pediátrico (ITP).
Resultados: Los accidentes de tráfico fueron la causa del traumatismo en el 27,8% de los pacientes, por detrás de las caídas casuales (41%); pero fue la causa del 38% de los pacientes con ITP=8 y el 70% de los pacientes con ITP=5. El 62% de los pacientes fueron varones. El 64% de los traumatismos ocurrieron en las ciudades, el 25% en carretera y el 11% en el campo. Los accidentes de automóvil fueron la causa más frecuente (39%), seguido de los atropellos (32%) y los accidentes de bici (29%). Se observa una curva bimodal en el rango de edades, siendo más frecuente en el período preescolar y entre los 10-14 años. El 24.5% presentaban un ITP ¾ 8 (trauma grave). A pesar de que en el RTP el aparato locomotor es el sistema más afectado (en el 51% de los accidentes), en los accidentes de tráfico es el craneofacial el sistema más vulnerable (75.6%). Un paciente falleció como consecuencia de un TCE severo. El RTP no incluye los pacientes fallecidos prehospitalariamente.
Conclusiones: Los accidentes de tráfico constituyen la tercera parte de los ingresos por traumatismo en niños. Su iincidencia aumenta exponencialmente al disminuir el ITP. Debido a su gran morbi-mortalidad deben someterse a especial reflexión para realizar mejoras en prevención primaria y secundaria, adecuación de los recursos sanitarios y corrección de deficiencias en el sistema de asistencia. 5000 niños menores de 15 años morirán en los próximos 10 años en España como consecuencia de accidentes de tráfico si no hacemos nada para evitarlo.
P259 CONSULTA EN URGENCIAS DE PEDIATRÍA POR CUERPO EXTRAÑO ALOJADO EN VÍA AERODIGESTIVA U OTRO ORIFICIO NATURAL. 10:25 h
Eva Gembero Esarte, Lourdes Gómez Gómez, Eva Rupérez García, Diana Martínez Cirauqui, Mercedes Herranz Aguirre, Alberto Pérez Martínez, Natividad Viguria Sánchez, Nuria Clerigué Arrieta
Hospital Virgen del Camino, Pamplona (Navarra).
Objetivos: Describir la epidemiología de los pacientes que consultaron en el Servicio de Urgencias de Pediatría del Hospital Virgen del Camino por ingestión, aspiración o introducción de un cuerpo extraño (CE) en cualquier orifico anatómico durante el año 2003.
Material y método: Estudio retrospectivo mediante revisión de las historias clínicas de aquellos pacientes cuyo motivo de consulta fue un accidente con CE, bien por ingestión-aspiración o por introduccción en algún orificio natural. Las variables analizadas mediante SPSS fueron: edad, sexo, mes del año, hora de consulta, si acudía remitido por otro faculatativo o de forma espontánea, las pruebas complementarias realizadas y el destino del paciente, así como la localización y tipo de CE. Se revisaron las intervenciones quirúrgicas (broncoscopia o esofagogastroscopia) por este motivo.
Resultados: Durante el año 2003 acuden 180 pacientes menores de 15 años (0,45% del total de pacientes atendidos) con la sospecha de ingesta-aspiración de CE o por haber introducido el CE en algún orificio anatómico. El 82% consulta por iniciativa propia. El 49% acude en el período de junio-septiembre. Si bien la distribución por edades no es significativa, el 46,6% está incluido entre 3 y 7 años. No existe predominio de sexo salvo en los casos de ingestión y aspiración en los que la relación niño/niña es 2/1 respectivamente. El 61% de los objetos están alojados en vía digestiva (37% en amigdalas, 12% esófago), 25% en nariz, 10% en oidos y 4% en bronquios. Los objetos son muy variados, si bien más del 50% de los CE ingeridos son monedas, y 1 de cada 2 CE aspirados son frutos secos. En 1 de cada 5 pacientes no se objetiva el CE. Cuando el CE está en oido, nariz o amigdala, en 1 de 4 casos se precisa otorrino para su extración. En los 7 pacientes con sospecha de aspiración de CE se realizó broncoscopia (en 2 no se encontró). El 18% precisaron esofagogastroscopia.
Conclusiones: La sospecha de un CE ingerido, aspirado o introducido en algún orificio es un motivo frecuente de consulta urgente en pediatría. La edad más frecuente, entre 3 y 7 años. Discreto predominio en varones. Predominan las monedas en la ingestión, y los frutos secos en la aspiración.
P260 "NO PATOLOGÍA URGENTE" EN PEDIATRÍA: ¿SOBREUTILIZACIÓN DE LOS SERVICIOS DE URGENCIAS?. 10:30 h
M. Nuria Fernández González, Josefina Fernández Antuña, José David Herrero Morin, Susana Parrondo Garrido, Pedro García Peleteiro, Antón Castaño Rivero
Hospital de Cabueñes, Gijón (Asturias).
Introducción y objetivos: Las urgencias pediátricas han aumentado de manera preocupante en los últimos años sin correlacionarse con el aumento de población infantil. Los casos sin patología objetivable suponen una proporción importante del número de visitas a servicios de urgencias pediátricas y una sobreutilización de los mismos. Se pretende describir las visitas pediátricas a urgencias sin enfermedad aparente que las justifique.
Material y métodos: Se revisan retrospectivamente las historias clínicas de todos los pacientes pediátricos vistos en nuestro servicio de Urgencias entre Julio y Diciembre de 2003 en los que no se encontró patología objetivable. Se recogen datos epidemiológicos, clínicos y asistenciales.
Resultados: Se revisan un total de 240 pacientes que suponen el 2,32% de las urgencias pediátricas atendidas en este período. Casi 3/4 partes de ellos son neonatos o lactantes (20,5% y 49,8% respectivamente), con leve predominio del sexo masculino. Acuden sobre todo en días laborables y durante el turno de tarde, siendo la irritabilidad el motivo de consulta más frecuente. Únicamente el 7,9% de ellos acude derivado desde su Centro de Salud. Se realizan exploraciones complementarias en 1/3 de los casos. El tiempo de espera hasta ser atendidos y total de estancia medios en urgencias son 33,39 y 105,15 minutos respectivamente. Son diagnosticados fundamentalmente por pediatras seguido de médicos de familia y residentes. Sólo el 15,4% de ellos presentan patología de base, destacando el asma como la más frecuente. La edad media materna es 30,8 años, encontrándose la mayoría de ellas entre los 31 y los 40 años. Los pacientes son hijos únicos en 2/3 de los casos.
Conclusiones: Los pacientes sin patología urgente vistos en nuestro servicio son fundamentalmente neonatos y lactantes, hijos únicos y sin patología de base. Suponen un porcentaje importante de las urgencias, con el consiguiente gasto sanitario. La mayor parte de estas consultas podrían ser evitadas con una mejor información familiar en puericultura básica.
P261 DIAGNÓSTICO DE ALERGIA EN UN SERVICIO DE URGENCIAS PEDIÁTRICO. 10:35 h
Eider Astobiza Beobide, Rocío Lamarca Gay, Nerea Trebolazabala Quirante, Pilar Galán del Río, Itziar Fernández Respaldiza, Cristina García Escudero, Aitziber Pérez Fernández, Amagoia Andrés Olaizola
Hospital de Cruces, Barakaldo (Vizcaya).
Objetivo: Diagnóstico definitivo de lactantes que presentando clínica de alergia en urgencias, fueron remitidos a las consultas externas de alergología.
Material y métodos: Estudio retrospectivo de todos los lactantes con sospecha de alergia valorados en nuestro servicio durante el 2002 y remitidos para identificación del alérgeno.
Resultados: De 127 episodios diagnosticados de alergia en nuestro servicio durante el 2002, se excluyeron 55 por no cumplir los todos los criterios. El 50% fueron mujeres y la edad media de 8,16 meses. Los motivos de consulta fueron: urticaria 40 (55,6%), exantema inespecífico 20 (27,8%) y angioedema 12 (16,6%) El diagnóstico más frecuente en urgencias fue la alergia alimentaria 59 casos (82%) y solo 10 casos se atribuyeron a medicamentos (13.9%) La mayoría de los niños no precisaron tto en la urgencia 61 (84.7%), precisando adrenalina intramuscular 9 casos (12.5%) Se pautó tto antihistamínico en 20 casos (16,7%) y la retirada del alérgenos sospechoso en 62 casos (86,2%) Un 20% de los niños presentaban cuadro infeccioso concomitante y un 16,7% tomaba algún medicamento por tal motivo. Preguntados los padres sobre si ellos relacionan el episodio con un desencadenante concreto 11 (15,2%) lo relacionan con antibiótico y el resto con algún alimento, sobretodo leche (51,3%) y huevo (8,3%) Un 13,9% era asmático y hasta un 26,4% presentaba dermatitis atópica. La proporción de los niños con alergia alimentaria es mayor en los niños con dermatitis atópica frente a los que no la tiene, 50% frente 8,5% respectivamente (p < 0,001) En 55,6% existían antecedentes familiares de asma o alergia. El diagnóstico se realizó mediante prick, rast, provocación o una combinación de varias. El prick fue positivo en 55,6% de los casos, el rast en 15,3% y la provocación 9,7%. En 61,8% casos se confirmó una alergia, siendo la más frecuente la alergia a la leche (50%)
Conclusiones: La mayoría de las alergias alimentarias, sobretodo en las etapas tempranas de la vida quedan confirmadas. La proporción de niños con alergia alimentaria es mayor en los niños con dermatitis atópica frente a los que no lo tienen. La alergia a medicamentos es poco habitual, tratándose en la mayoría de las ocasiones de exantemas producidos por el propio cuadro infeccioso.
P262 ESTUDIO CLÍNICO DESCRIPTIVO DE LA CAMPAÑA DE PREVENCIÓN DE BRONQUIOLITIS AGUDA EN NUESTRO HOSPITAL: PERÍODO 2002-2003. 10:40 h
Intzane Ocio Ocio, Beatriz Rodríguez Pérez, Antoni Matilla Fernández, Iratxe Salcedo Pacheco, Gontzal Martínez de la Hidalga Ortiz de Zarate, Concepción Salado Marín, Enrique González Molina, M. Ángeles Fernández Cuesta, Alfredo Bosque Zabala, M. Arnaiz Uyarra
Hospital Txagorritxu, Vitoria (Álava).
Introducción: La bronquiolitis aguda es básicamente una enfermedad del lactante, motivo frecuente de consulta en los servicios de urgencias pediátricas y una de las principales causas de hospitalización en niños menores de un año. Presentamos los datos de un estudio descriptivo de los cuadros de bronquiolitis atendidos en nuestra urgencia, desde octubre del 2002 a marzo de 2003, continuando con la campaña de prevención de bronquiolitis iniciada en octubre de 2000 y dirigida desde la maternidad a las familias de los niños nacidos en nuestro hospital.
Material y métodos: definimos bronquiolitis aguda como el primer episodio de broncospasmo en lactante menor de 18 meses. El estudio se realizó mediante encuesta individualizada a los pacientes atendidos en nuestro servicio de urgencias por bronquiolitis. Los datos se recogieron en el momento de la asistencia y estaban en relación con el proceso agudo, antecedentes de riesgo personales y familiares y datos de la campaña de prevención y detección del antígeno VRS en muestra de secreción nasal.
Resultados: Durante el período de estudio se atendieron 11190 urgencias,3269 correspondieron a lactantes menores de 18 meses, de los cuales 191 (17,2%) fueron diagnosticados de bronquiolitis. El pico de máxima incidencia de bronquiolitis correspondió a noviembre y diciembre con un 59,2%. Se obtuvieron muestras para VRS en 162 (84,8%) y en 78 (48,1%) resultaron VRS (+). Se observa que el pico de máxima incidencia de bronquiolitis coincide con un mayor porcentaje de positividad para el VRS (35,9% en noviembre y 41% en diciembre). El 46,6% presentaba antecedentes personales o familiares de alergia/atopia. El 53,4% tabaquismo familiar. En el 69,6% tenían ambiente familiar catarral y en el 52,4% tenían hermanos menores de 5 años. El 6,8% habían presentado patología respiratoria en la etapa neonatal. No se encontraron diferencias significativas en relación al sexo. Ingresaron el 20,4% (39), el 23,3% precisó oxigeno durante su ingreso. En el 58,1% de los casos ingresados se administró beta2 y en el 48,4% adrenalina nebulizada, corticoides sistemicos en el 22,5% y antibióticos en el 58,1%, que puede relacionarse con una mayor incidencia de patología asociada (35,1%). Se valoran los días de ingreso en relación al tratamiento recibido.El 42% confirmaron haber recibido información de la campaña de prevención de infecciones respirato
P264 UTILIZACIÓN DE MATERIAL SEMIRÍGIDO PARA EL TRATAMIENTO DEL TRAUMATISMO PERIFÉRICO. 10:50 h
Susana Capapé Zache, Nerea Trebolazabala Quirante, Ana Fernández Landaluce, Elena Mora González, Beatriz Azkunaga Santibañez, Javier Benito Fernández, Santiago Mintegui Raso, Jesús Sánchez Etxaniz, Miguel Ángel Vázquez Ronco
Hospital de Cruces, Barakaldo (Vizcaya).
Objetivo: Describir nuestra experiencia en el uso de material semirígido para la inmovilización de extremidades en pacientes pediátricos.
Material y método: Estudio prospectivo de 200 traumatismos periféricos en los que se realizó inmovilización con material semirígido, recogiendose datos sobre la inmovilización y control posterior a las 2 semanas en el servicio de urgencias.
Resultados: De los 200 episodios recogidos, 29 se desecharon por no acudir a control. De los 171 niños controlados 46,8% fueron varones y la edad media 9,5 años. En todos los niños se realizó radiografía excepto en 2 esguinces de tobillo, que se inmovilizaron directamente. Fueron normales todas las radiografías excepto una (fractura diafisaria de tibia) El traumatismo tobillo/pierna fue el diagnóstico más frecuente, 91 casos, seguido de antebrazo/muñeca 43 y dedos 36, un caso se diagnosticó de fractura de tibia. Se realizó inmovilización de tobillo/pierna en 92 pacientes (53,8%) de muñeca/antebrazo 43 (25,1%) y de dedos 36 (21,1%). Todos los niños fueron valorados por el pediatra y uno de ellos conjuntamente con traumatología. La mayoria de los niños, 146 (85,4%), fueron inmovilizados por el médico adjunto, 23 (13,5%) por residentes y 2 (1,2%) por enfermería. La evolución fue favorable en la mayoría de los casos, 143 niños (83,6%) en el control realizado a las 2 semanas. Persistía dolor en 28 niños realizándose segunda radiografía en 24 de ellos (85,7%) resultando 2 alteradas (fisuras dedo pie y mano). En 4 casos no se realizó control radiológico, en 3 de ellos persistía dolor leve que se trató con AINEs y uno (trauma tobillo) se remitió a traumatología. De los 22 restantes, 9 fueron remitidos para valoración por traumatólogo; 13 continuaron con inmovilización 1 semana más. De estos niños, 10 fueron controlados en urgencias y 3 por su pediatra, todos con evolución favorable.
Comentarios: La valoración inicial de los niños con traumatismo periférico debe realizarse en primer lugar por el pediatra de urgencias. En nuestra experiencia creemos que la utilización de material semirígido para el tratamiento de este tipo de lesiones de extremidades es una buena opción, dado que la mayoría de los niños tienen una evolución favorable.
P265 INTOXICACIÓN POR INGESTA ACCIDENTAL DE CANNABIS EN UN VARÓN DE 20 MESES CON ALTERACIÓN AGUDA DEL NIVEL DE CONCIENCIA. 10:55 h
Pedro J. Alcalá Minagorre, Juan Utrero Sánchez, Esther Blanco Alemany, María González Santacruz, Eduardo Martínez Salcedo, M. del Carmen Vicent Castelló
Hospital General Universitario, Alicante.
Observación clínica: Varón de 20 meses remitido desde otro centro sanitario por disminución de conciencia de instauración brusca. En su hospital de origen se objetivó un nivel de conciencia de 9 (según escala de Glasgow modificada para lactantes), con pupilas midriáticas escasamente reactivas, sin otros signos de focalidad neurológica. Se le realizó un hemograma, bioquímica sanguínea, TC craneal y punción lumbar, resultando todos ellos normales. Ante la persistencia de la clínica y la posible necesidad de ventilación mecánica, se decidió su traslado a nuestro centro hospitalario. A su llegada el paciente se mostraba hemodinámicamente estable, con respiración espontánea, manteniendo inicialmente la disminución del nivel conciencia. Pese a que los padres negaron la posibilidad de que el niño estuviera en contacto con cualquier tipo fármaco o de droga legal/ilegal, se detectó la presencia de cannabis en tres muestras consecutivas de orina. Finalmente, reconocieron que el niño había ingerido un fragmento de pastilla de hachís previo al inicio de los síntomas. El paciente evolucionó favorablemente, recuperando el nivel de conciencia normal a las 12 horas de iniciarse el cuadro. El caso fue notificado a las autoridades judiciales.
Comentarios: El importante consumo de cannabis en la sociedad actual facilita el acceso de los pacientes pediátricos a esta droga. En niños pequeños la intoxicación se produce generalmente por la ingestión accidental. Los efectos por vía oral son más lentos y duraderos que por la inhalatoria. La sintomatología comprende las náuseas y vómitos, sequedad de boca, hiperemia conjuntival, palidez, y alteraciones neuroconductuales. En ocasiones el cuadro puede alcanzar una gravedad considerable: coma con depresión respiratoria y necesidad de ventilación mecánica, bradicardia y/o hipoglucemia. El tratamiento de la intoxicación aguda incluye el lavado gástrico, el carbón activado y medidas de soporte. Se ha descrito la utilización exitosa de flumazenilo en casos de coma tras consumo de cannabis. Ante la sospecha clínica fundada, se debe realizar siempre una determinación de tóxicos en orina del paciente, pese a que los padres o acompañantes nieguen la posibilidad de un contacto con fármacos o substancias ilegales.
P266 ATAXIA DE APARICIÓN AGUDA: ETIOLOGÍA, MANEJO Y SEGUIMIENTO. 11:00 h
Sonia Martínez González, Eider Astobiza Beobide, Mª Jesús Martínez González, Ainhoa García Ribes, Santiago Mintegui Raso, Javier Benito Fernández, José M. Prats Viñas
Hospital de Cruces, Barakaldo (Vizcaya).
Objetivo: Describir la etiología, actitud diagnóstico-terapeútica, y seguimiento de los niños que consultaron por ataxia de aparición aguda en Urgencias de Pediatría.
Pacientes y métodos: Estudio retrospectivo de los 38 niños diagnosticados de ataxia aguda en nuestro hospital durante los últimos 3 años (2001-2003).
Resultados: Durante este período se valoraron 159.002 episodios en Urgencias, 38 niños (0,023%) presentaron una ataxia aguda. Las causas más frecuentes fueron: 1) Postinfecciosa 18 (47,3%). La edad media de estos niños fue de 55,7 ± 27,89 meses, siendo un 55,5% entre 2 y 4 años, y un 57% niñas. El tiempo de evolución del cuadro hasta la aparición de la ataxia fue de 136 ± 86,33 horas. La etiología fue: varicela (9), viral inespecífica (6), micoplasma (1), enterovirus (1), y EBV (1). Se realizaron estudios complementarios al 89%. El examen del LCR fue patológico en 10 de 15 casos. La neuroimagen fue normal. Precisaron ingreso 14 (77,7%), con una duración media de 5,14 ± 2,98 días. La exploración neurológica al alta persistía alterada en 12. Todos se recuperaron en su totalidad, salvo un paciente, que debutó así con una linfohistiocitosis hemofagocítica. 2.- Intoxicación 10 (26,3%). Este grupo supone un 1.8% del total de consultas por ingesta de tóxicos. La edad media fue de 39,55 ± 40,42 meses, con predominio de varones (67,6%). Los fármacos fueron benzodiacepinas (4), anticatarrales (3), fenobarbital, sobredosificación de fenitoina, y etanol. El tiempo de evolución fue de 2,4 ± 1,6 horas, y la clínica más frecuente, vómitos y somnolencia. Ninguno preciso ingreso ni seguimiento neurológico. 3.- TCE 2 (5,2%). 4.- Miscelánea 8 (21%), grupo heterogéneo que englobaba: vértigo paroxístico (2), encefalomielitis aguda diseminada, tumor paravertebral, Guillain-Barré atípico, ataxia en episodio febril de un niño con síndrome de desviación paroxística de la mirada, ataxia funcional e idiopática.
Conclusiones: Las ataxias agudas son un motivo de consulta infrecuente en Urgencias de Pediatría. Las ataxias agudas postinfecciosas y por intoxicación son las más usuales, siiguiendo por lo general un curso benigno y autolimitado. La historia clínica y la exploración neurológica nos orientaran hacia la etiología, y ésta al uso adecuado de los estudios complementarios. La neuroimagen y el ingreso hospitalario debieran reservarse para presentaciones atípicas, signos de focalidad neurológica, y duración prolongada del cuadro.
P267 EFECTO DE LA APLICACIÓN DE UN PROTOCOLO DIAGNÓSTICO SOBRE LA ADECUACIÓN DEL USO DE LA RADIOGRAFÍA EN EL TRAUMATISMO CRANEOENCEFÁLICO PEDIÁTRICO. 11:05 h
Pedro J. Alcalá Minagorre, Jesús Aranaz Andrés, José Flores Serrano, Laura Asensio García, Amelia Herrero Galiana, Eduardo Martínez Salcedo
Hospital General Universitario, Alicante y Universidad Miguel Hernández, San Juan de Alicante (Alicante).
Antecedentes: El traumatismo craneal constituye un motivo frecuente de consulta en Urgencias. La utilización de las pruebas de imagen, especialmente la radiografía, está sujeta a una importante variabilidad en la práctica clínica, pudiendo repercutir en la calidad asistencial.
Objetivo: Estimar la idoneidad del uso de la radiografía de cráneo antes y después de la aplicación de un protocolo diagnóstico en niños (0-14 años) que acuden a urgencias con traumatismo craneoencefálico durante período de 24 meses.
Métodos: Audit clínico, pretest-postest. Consta de dos fases, cada una de ellas de 12 meses de duración: 1) Estudio descriptivo retrospectivo sobre el uso y determinantes de la radiografía de cráneo en los traumatismos craneoencefálicos. 2) Estudio de intervención para evaluar el uso de la radiografía de cráneo tras la aplicación de un protocolo diagnóstico, basado en las recomendaciones disponibles en la bibliografía, consensuado y validado.
Resultados: Se recogieron 1362 traumatismos craneales (1,71% del total de urgencias pediátricas atendidas). La utilización de la radiografía se redujo significativamente, pasando del 45,8% al 38,9% tras la aplicación del protocolo. Así también se produjo una disminución significativa de la inadecuación (conjunto de infrautilización y sobreutilización) de la prueba, pasando del 25,1% al 14,7%. La sobreutilización pasó del 17,3% al 11,3%, y la infrautilización del 7,8% al 3,4%. No hubo diferencias en la utilización de la tomografía computerizada craneal, y sí se produjo una disminución significativa de los ingresos.
Conclusiones: La elaboración y aplicación de un protocolo de actuación basado en las recomendaciones vigentes, con participación interdisciplinar y adaptada a las necesidades asistenciales, mejora la calidad asistencial, la precisión diagnóstica y el consumo de recursos sanitarios. En este caso, no sólo se redujo el número total de radiografías solicitadas, sino que su utilización se adecuó mejor a la situación clínica del paciente. De todos modos, los protocolos, si no se combinan con la definición de procesos, el apoyo de decisiones, y una adecuada difusión, difícilmente disminuyen la variabilidad en la práctica clínica.
P268 QUISTE EPIDERMIODE INTRACRANEAL. UN HALLAZGO CASUAL EN LA SALA DE URGENCIAS. 11:10 h
Esther Vázquez López, Francisco Javier González Gómez, Ana García González, Alba Manjón Herrero, M. Isabel López Conde, Soledad Martínez Regueira, Ramón Morales Redondo
Complexo Hospitalario Xeral-Calde, Lugo.
El quiste epidermoide craneal es una entidad rara en la infancia. Constituye menos del 1% del total de los tumores craneales. El 25% de los quistes epidermoides son intradiploides y el 75% son intradurales. Se caracteriza por tratarse de una lesión lítica bien definida y con bordes escleróticos. El diagnóstico se produce a veces como un hallazgo casual al realizar una radigrafía de craneo por otros motivos. Su diagnóstico de confirmación es a través de una biopsia, aunque su imagen radiológica es tremendamente sugestiva. El tratamiento es quirúrgico (exéresis). Su pronóstico es favorable y su recurrencia es excepcional. Ante un hallazgo radiológico de este tipo siempre hemos de plantearnos un diagnóstico diferencial con otras entidades como una histiocitosis de células de Langerhans, granuloma eosinófilo, lesión metastásica...
Presentamos el caso de una niña de 23 meses de edad que acude a urgencias tras sufrir un traumatismo creaneoencefálico con perdida de conciencia de unos segundos de duranción. Su exploración física era normal, salvo una pequeña masa dura que se palpaba a nivel fronto-parietal izquierdo. Se solicita una radiografía de cráneo y como hallazgo casual nos encontramos con una lesión lítica de bordes bien definidos de unos 3 cm de diámetro a nivel frontal izquierdo compatible en primer lugar con un quiste epidermoide craneal. En el TAC craneal se describe a esta lesión como una lesión lítica y destructiva, en la unión frontoparietotemporal izquierda de 1,3 x 0,8 cm de diámetro. Se trata de una lesión lítica expansiva con destrucción de la tabla ósea interna y externa y sin afectación intracraneal. Ante la sospecha de quiste epidermoide se ingresa a la paciente para realizar un adecuado diagnóstico diferencial. Se realizó una bioquímica sérica completa que resultó ser normal, al igual que una serie ósea, una ecografía abdominal y un EEG.
Es interesante tener en mente una serie de imágenes radiográficas que podemos encontrar de forma casual en una sala de urgencias para que nos permita hacer una orientación diagnóstica rápida y no pasen desapercibidas aunque no sean el motivo principal de consulta.
P269 EPIDEMIOLOGÍA DE LOS ACCIDENTES INFANTILES EN SALAMANCA EN EL AÑO 2002. 11:15 h
Susana Grande Bárez, Antonio Grande Benito, Froilan Hidalgo Acera, Jesús Prieto Veiga, Ana Periañez Marchal, Olga González Calderón, Carmen Rubio Álvarez, Ana Gloria Andrés Andrés, Alfonso Rodríguez Albarrán
Hospital Universitario, Salamanca.
Las lesiones por accidente son la primera causa de muerte o discapacidad y una importante fuente de morbilidad en la población infantil mayor de un año de los países desarrollados. Desde el 1 de enero hasta el 31 de diciembre de 2002, hemos realizado un estudio retrospectivo de todos los niños menores de 14 años que consultaron por accidentes en los Servicios de Urgencias del Hospital Universitario de Salamanca. Durante el mismo período realizamos un seguimiento diario de los pacientes menores de 14 años que precisaron hospitalización por accidente.
De las 22.140 urgencias infantiles, 5.327 (24,06 %) fueron accidentes, y de los 2.151 ingresos pediátricos (excluyendo neonatos), 169 (7,86 %) fueron por accidente. No se han podido incorporar datos de 180 pacientes, por lo que el estudio está realizado sobre 5.147 niños, de los cuales, 4.978 (96,72%) son derivados a su domicilio tras ser atendidos en los Servicios de Urgencias y 169 (3,28%) ingresan. La edad media es de 7,49 años. El 43,02% fueron mujeres y el 56,98% varones. El mes con mayor número de accidentes fue Mayo, con 495, y el mes con mayor número de ingresos, Agosto con 27. El día con más consultas por accidentes fueron los lunes (806 casos). El horario en el que más niños acuden a urgencias por un accidente es de 17:00 a 20:59 horas (1.719 casos), siendo también el período con mayor número de ingresos (62). La causa más frecuente de accidentes fueron los traumatismos (caídas, accidentes de tráfico, deportivos, heridas...etc.) con 4.637 casos, seguidos de los 230 casos de cuerpos extraños, 113 intoxicaciones, 83 lesiones por animales y 62 quemaduras. El 60% de accidentes (3.081) se producen en la calle; el 25% (1.287) en el domicilio y el 15% (779) en el colegio. Fallecen 3 de los niños accidentados atendidos en el hospital, uno por accidente de tráfico, otro por accidente deportivo (caída de portería de fútbol sobre el niño) y otro por intoxicación por monóxido de carbono. Reciben atención médica prehospitalaria por equipos de atención primaria o unidades móviles de soporte vital (básico y avanzado), 651 niños de los que no precisan ingreso y 74 de que ingresaron. La epidemiología de los accidentes infantiles en nuestro medio es similar a la de otros estudios; es fundamental conocer dicha epidemiología para llevar a cabo estrategias de prevención dirigidas a disminuir la morbi-mortalidad por accidente.
P270 CARACTERÍSTICAS CLÍNICAS Y EPIDEMIOLÓGICAS DE LOS PACIENTES INGRESADOS EN SALA DE OBSERVACIÓN DE UN HOSPITAL TERCIARIO EN UN PERÍODO DE DOS AÑOS. 11:20 h
Eva Gembero Esarte, Mercedes Herranz Aguirre, Marta González Villar, Beatriz Solís Gómez, Nuria Clerigué Arrieta, Eva Rupérez García, Natividad Viguria Sánchez, Lourdes Gómez Gómez, Clara Lezaun Lecea, Mercedes Galar Jiménez
Hospital Virgen del Camino, Pamplona (Navarra).
Antecedentes y objetivos: Describir la utilización de la sala de observación, las características clínicas y epidemiológicas de los pacientes ingresados en esta unidad y su destino al alta.
Métodos: Estudio observacional descriptivo realizado en el Servicio de Urgencias Pediátricas de un Hospital terciario, sobre una muestra de 2.438 pacientes de un total de 74.989 que acudieron al Servicio de Urgencias en un período de dos años(1-6-01/ 31-5-03). Para ello, seleccionamos a los pacientes mediante un muestreo consecutivo de todos los ingresados en la sala de observación durante el período del estudio. Se registraron las horas de ingreso y alta, duración de la estancia, edad, pruebas complementarias realizadas, tratamientos pautados, diagnóstico al alta y destino de cada paciente. Los datos recogidos fueron analizados con el programa estadístico SPSS para windows.
Resultados: Del total de pacientes vistos en el Servicio de Urgencias durante el período del estudio ingresaron en la sala de Observación un 3,25%, con una estancia media de 5,5 horas. La edad media fue de 5,3 años. El análisis de sangre y la radiología fueron las pruebas complementarias más empleadas y la sueroterapia y los antitérmicos los tratamientos más pautados. La patología más frecuente fue la digestiva (diarrea/gastroenteritis, vómitos), representando el 29,98%, seguido de patología respiratoria (bronquitis/asma, laringitis), 12,42%, fiebre 9,84%, TCE 8,73%, convulsiones 8,57% e intoxicaciones 5,16%. Los destinos definitivos fueron: domicilio 74,5%, ingresos 25,3% y otros 0,2%.
Conclusiones:1) El 3,25% de los pacientes que consultaron en urgencias precisaron esta unidad. 2) El destino definitivo al alta fue en un 75,5% su domicilio. 3) La estancia media de los pacientes en nuestra unidad fue corta (5,5 horas). 4) La patología atendida en sala de observacion, ademas de ser la mas frecuente en los niños, es aquella que puede experimentar cambios clínicos significativos en un período breve de tiempo, y por tanto es susceptible de vigilancia y tratamiento en una unidad de este tipo.
P271 VALORACIÓN DEL LACTANTE FEBRIL MENOR DE UN MES. 11:25 h
Yolanda María Chica Fuentes, José Manuel Jiménez Hinojosa, Olga M. Escobosa Sánchez, Vanessa Alonso Morales, M. Pilar Ranchal Pérez, Antonio Madrid Madrid, Isabel Durán Hidalgo, Custodio Calvo Macías, Antonio Jurado Ortiz
Hospital Materno Infantil Carlos Haya, Málaga.
Introducción: El riesgo de padecer infección bacteriana grave (IBG) sin clínica está en relación inversa con la edad. Los neonatos presentan escasa expresividad clínica y el grado de fiebre no predice la gravedad de su enfermedad.
Objetivo: Valorar la actitud clínico-terapeútica ante estos pacientes.
Material y métodos: Estudio retrospectivo de los pacientes < 1 mes diagnosticados de fiebre sin foco (FSF) en nuestro servicio de urgencias, entre el 1/01/02 y el 31/12/02. Se analiza su evolución y la incidencia de IBG para determinar la necesidad de realizar o no screening de sepsis y posterior tratamiento.
Resultados: Durante este período se atendieron un total de 2500 niños < 3 años con FSF, de estos 127 (5%) eran lactantes < 1 mes, con una media de edad de 21 días. Todos ellos presentaban una temperatura rectal superior a 38ºC y un tiempo de evolución inferior a 72 horas. A su llegada se realiza una historia y exploración física minuciosa, y un screening infeccioso consistente en hemograma, PCR, procalcitonina, hemocultivo, sedimento urinario y urocultivo, por sondaje vesical, y citoquímica y cultivo de LCR. Independientemente de los resultados del screening todos fueron hospitalizados, pero 6 (4.7%) precisaron ingreso en UCIP, 91 (71%) en planta y 30 (23%) en observación. De los 6 pacientes ingresados en UCIP hubo 4 sepsis con hemocultivo positivo (2 E. coli y 2 S. agalactie), 4 urocultivos positivos (3 E. coli y 1 Klebsiella) y 2 meningitis con cultivo de LCR positivo (1 E. coli y 1 S. agalactie). Entre los lactantes ingresados en planta hubo 38 infecciones de orina con urocultivo positivo y 1 celulitis, el resto fueron infecciones virales. Los pacientes que quedaron en observación fueron dados de alta después de un período de 12 horas, por detectarse algún síntoma respiratorio o de otro tipo, y ninguno ha presentó complicaciones. En conjunto, el 35.4% de los lactantes ingresados tenían una IBG.
Conclusiones: Dada la alta incidencia de IBG en estos niños y de acuerdo con la evidencia científica, debe realizarse screening de sepsis e ingreso para tratamiento antibiótico intravenoso en espera de cultivos en todos los lactantes < 1mes con FSF. Los estudios que investigan la posibilidad de tratamiento ambulatorio en pacientes clasificados como de bajo riesgo, muestran un 5-10% de IBG en este grupo. Ni la exploración ni las pruebas complementarias son seguras para evaluarlos.
P272 ¿QUÉ INGRESAMOS EN EL HOSPITAL PEDIÁTRICO DE CORTA ESTANCIA?. 11:30 h
Bárbara Fernández Barrio, Rocio Quiroga Gonzalez, Sonsoles Suárez Saavedra, Alejandro Pérez Guirado, Amparo Calvo Gómez-Rodulfo, José Luis Fanjul Fernández, José Andrés Concha Torre
Hospital Universitario Central de Asturias, Oviedo (Asturias).
Antecedentes y objetivos: El Hospital Pediátrico de Corta Estancia (HPCE) de nuestro Departamento consta de 6 camas y funciona para observación y tratamiento de niños que inicialmente no requieren ingreso en planta. El objetivo del estudio es conocer las características de los pacientes y las patologías que ingresan en el HPCE; determinar las estancias prolongadas y las derivaciones a hospitalización.
Material y métodos: Estudio retrospectivo, descriptivo de los ingresos en el HPCE durante 4 años (enero-00 a diciembre-03). Datos obtenidos del registro de ingresos e historias clínicas (edad, sexo, servicio responsable, tipo de patología, día, turno y mes de ingreso, estancia y evolución). Los resultados se almacenaron en una base de datos en el programa SPSS y se realizó análisis estadístico de los mismos.
Resultados: Ingresaron en el HPCE en el período de estudio 5.375 pacientes (3,67 pacientes/día) con edad media de 60,45 ± 50,3 meses y predominio de varones (60%). Los ingresos fueron significativamente más frecuentes en turno de tarde (41,5%) y los días de menos ingresos fueron sábados y domingos, en relación inversa a la asistencia a Urgencias. ? partes de los ingresos fueron realizados por Pediatría y un 18% por Cirugía Pediátrica. Respecto a los ingresos del Servicio de Pediatría, un 23,4% correspondió a procesos digestivos, el 13,7% a procesos infecciosos que incluían fiebre sin foco y el 11,9% a procesos respiratorios. Se apreció disminución de los ingresos en meses de verano en relación con menor frecuentación a Urgencias. La estancia media fue de 17,6 ± 11,8 horas con un 9,6% de pacientes con estancia superior a 36 horas. El 80,6% de pacientes fueron dados de alta a domicilio con un porcentaje significativo (18,6%) que fue trasladado a planta de hospitalización, quirófano o cuidados intensivos.
Comentarios: El HPCE es una herramienta útil para la observación y tratamiento de pacientes sin patologías graves en las primeras horas de evolución de su enfermedad. En nuestro estudio, el número de pacientes con estancia prolongada (superior a 36 horas) no es elevado e indica una adecuada rotación de las camas. El porcentaje de pacientes que se trasladan desde el HPCE a hospitalización es alto, lo que indica una inadecuada utilización del mismo para determinadas patologías. El establecimiento de criterios de ingreso para el HPCE y vías clínicas para algunas patologías permitirá reducir el número de estancias inapropiadas.
P273 PRIAPISMO: UN MOTIVO DE CONSULTA EXCEPCIONAL EN LA URGENCIA PEDIÁTRICA. 11:35 h
M. Luisa Herreros Fernández, Ana M. Pastor Gómez, Victoria Gómez dos Santos, Juana Barja Tur, Ricardo Díez García, Alfonso González Laguillo
Clínica Moncloa, Madrid.
Antecedentes y objetivos: El priapismo es una erección prolongada y dolorosa, siendo un motivo excepcional de consulta. La distinción entre sus dos tipos básicos, el de alto y el de bajo flujo, es esencial por su diferente tratamiento y pronóstico. Presentamos dos casos vistos en nuestra urgencia en el último año y realizamos una revisión de la literatura estableciendo un algoritmo de diagnóstico y tratamiento.
Métodos:Caso 1: Varón de 6 años de edad de origen Peruano con erección dolorosa de 2 horas sin otros síntomas asociados. En tratamiento actual con desmopresina y clormipramina por eneuresis nocturna. En la exploración se objetiva tumefacción dolorosa de cuerpos cavernosos sin signos de isquemia. Las exploraciones complementarias descartan patología hematológica. Se instaura tratamiento con metamizol iv con resolución del cuadro. Caso 2: Varón de 4 años de edad sin antecedentes de interés que consulta por erección dolorosa y retención urinaria de 12 horas de evolución. En la exploración presenta erección media con tumefacción dolorosa de cuerpos cavernosos sin signos de isquemia. Descartamos patología hematológica y se confirma infección urinaria por E. coli. El cuadro se resuelve tras tratamiento con metamizol iv.
Conclusiones: El priapismo es una consulta excepcional en la urgencia pediátrica. El adecuado enfoque diagnóstico y la distinción entre sus dos tipos básicos es esencial por su diferente tratamiento y pronóstico. La historia clínica, la exploración y el hemograma son el primer escalón diagnóstico. El ecodoppler peneano, la gasometría de cuerpos cavernosos y la arteriografía de iliacas permiten el diagnóstico definitivo.
P274 CARACTERÍSTICAS DEL LACTANTE DE 3-24 MESES QUE CONSULTA POR FIEBRE SIN FOCALIDAD (FSF) EN UN SERVICIO DE URGENCIAS PEDIÁTRICO HOSPITALARIO. 11:40 h
Aitziber Pérez Fernández, María González Balenciaga, Susana Capapé Zache, Nerea Trebolazabala Quirante, Elena Mora González, Ana Fernández Landaluce, Jesús Sánchez Etxaniz, Miguel Ángel Vázquez Ronco, Javier Benito Fernández, Santiago Mintegui Raso
Hospital de Cruces, Barakaldo (Vizcaya).
Objetivo: Conocer las características de los pacientes de 3-24 meses que consultan por fiebre sin focalidad (FSF) en un Servicio de Urgencias Pediátrico Hospitalario, el manejo en el mismo y su evolución posterior.
Pacientes y método: Revisión retrospectiva de todos los episodios correspondientes a lactantes de 3-24 meses de edad que consultaron en el Servicio de Urgencias Pediátrico del Hospital de Cruces por presentar fiebre sin focalidad entre 01-09-03 y 31-12-03.
Resultados: Durante ese espacio de tiempo se registraron en Urgencias 2755 episodios correspondientes a niños de 3-24 meses que consultan por fiebre. De éstos, 734 (26,6%) correspondieron a lactantes con fiebre sin focalidad (52.2% varones). 237 (32.3%) consultaron por cuadros de una duración ≤ 6 horas. Se practicaron exploraciones complementarias a 552 niños (75.2%), principalmente tira reactiva de orina (522 niños). En 121 ocasiones (16,4%) se practicó analítica sanguínea. Diagnósticos finales: 678 síndrome febril sin focalidad (SFSF), 53 ITU, meningitis 3. De los 678 diagnosticados en Urgencias de SFSF, 673 (99,2%) se manejaron de manera ambulatoria contactándose telefónicamente con 579 (86,0%). De éstos, 159 (27,4%) recibió posteriormente un diagnóstico diferente (destacando 3 neumonías, 3 ITU y una meningitis). El porcentaje de pacientes en los que el diagnóstico final varió fue superior en los que reconsultaron en el Hospital (58/93, 62.4%) que en los niños manejados posteriormente de forma exclusiva por su pediatra (101/486, 20,8%, p < 0,00001). Todos evolucionaron bien.
Comentarios: la consulta precoz de los lactantes de 3-24 meses con FSF en un Servicio de Urgencias Pediátrico Hospitalario dificulta el llegar inicialmente al diagnóstico definitivo de estos pacientes, por lo que la observación de los mismos, ya sea intrahospitalaria o en domicilio según los casos, es imprescindible independientemente del juicio clínico inicial.
P275 ESTUDIO COMPARATIVO DE LOS CASOS DE BRONQUIOLITIS DURANTE LAS CAMPAÑAS 2000-2001, 2001-2002 Y 2002-2003. 11:45 h
Beatriz Rodríguez Pérez, Intzane Ocio Ocio, Iratxe Salcedo Pacheco, Antoni Matilla Fernández, Gontzal Martínez de la Hidalga Ortiz de Zarate, Concepción Salado Marín, Enrique González Molina, E. Tato Eguren, Juan I. Montiano Jorge, Idoia Martínez Fernández de Pinedo
Hospital Txagorritxu, Vitoria (Álava).
Introducción: Presentamos los datos de un estudio comparativo de los casos de bronquiolitis atendidos en nuestras urgencias durante las campañas de octubre a marzo de 2000-2001,2001-2002 y 2002-2003 y las campañas de prevención de bronquiolitis realizadas desde nuestra área de maternidad a todas las familias que tuvieron un niño durante el período de estudio.
Material y métodos: Recogida de datos mediante encuesta individualizada en urgencias a los diagnósticos de bronquiolitis. Estudio descriptivo de cada período comparando la efectividad de la campaña, los datos epidemiológicos y terapéuticos.
Resultados: Se observa un incremento en el número de casos atendidos por bronquiolitis en relación con el segundo período, así como en el número global de urgencias (1,7%). Durante el período 00-01 el 1,96% de nuestras urgencias fueron diagnosticadas de bronquiolitis, frente al 1,42% en el período 01-02 y el 1,7% correspondiente al 02-03. Se evidencia además un cambio del pico de bronquiolitis, adelantándose con respecto a los períodos previos (noviembre-diciembre), siendo muy similar el porcentaje total de bronquiolitis atendidos en estos meses. Ha disminuido significativamente la recogida de muestra para VRS en la última campaña (00-01: 90,4%;01-02: 96,2%; 02-03: 84.8%) observando, a pesar de ello, un ligero incremento en el número de VRS(+) (00-01:71%; 01-02: 41,9%; 02-03: 48,1%). Se aprecia una disminución progresiva del número de padres que han recibido información, en relación a los años previos, manteniéndose el area de maternidad como principal fuente de información. Los parámetros asistencia a guardería, antecedentes de alergia/atopia, hermanos < 5años, tabaquismo materno y catarro en la familia muestran una correlación significativa en el desarrollo de bronquiolitis.En los niños que precisaron ser hospitalizados llama la atención que continúan disminuyendo los casos que precisan oxigenoterapia, beta2, adrenalina y corticoides sistémicos, aunque se evidencia una mayor necesidad de corticoides inhalados y antibioterapia; en relación, fundamentalmente, a una mayor incidencia de patología asociada en el momento del ingreso (00-01: 16%; 01-02: 33%; 02-03: 35,1%). Aún así, no se observan hospitalizaciones más prolongadas ni curso clínico más severo. Ha disminuído significativamente el porcentaje de ingresos (00-01: 23,7%; 01-02: 31,4%; 02-03: 16,3%)
P277 MANEJO DEL SÍNCOPE VASO-VAGAL EN URGENCIAS DE PEDIATRÍA. 11:55 h
Iñaki Ruiz Manzanal, Pilar Galán del Río, Susana Capapé Zache, Nerea Trebolazabala Quirante, Elena Mora González, Jesús Sánchez Etxaniz, Miguel Ángel Vázquez Ronco
Hospital de Cruces, Barakaldo (Vizcaya).
Objetivo: Conocer las características clínicas y manejo posterior de los niños diagnosticados de síncope vaso-vagal en el Servicio de Urgencias de Pediatría de un hospital terciario.
Material y metodo: Estudiamos de forma retrospectiva los 151 episodios con el diagnóstico de salida de sincope vaso-vagal durante los años 2001 y 2002. Se recogió información sobre epidemiología, manejo en la urgencia y necesidad de valoración por un especialista.
Resultados: La edad media fue de 105,4±39 meses. El 55,6% (84) fueron mujeres. Dos niños tenian antecedente de epilepsia y 2 cardiológico (estenosis pulmonar). El 76,8% (115) referían síntomas presincopales. La duración fue menor de un minuto en el 84,5% de los casos. Tras la recuperación refirieron somnolencia o confusión 12 niños (7,9%). La exploración física fue normal en todos los niños salvo en uno que se encontró un soplo cardíaco antes no diagnosticado. Se realizaron 50 (33,1%) glucemias capilares y se tomó la tensión arterial al 64,9%, siendo todas ellas normales. Solo se encontró una alteración electrocardiográfica (un marcapasos migratorio) del total de 100 (66,2%) que se realizaron. Electroencefalograma se realizó en 29 (19,2%) ocasiones encontrando alteración en un niño ya diagnosticado previamente (por este episodio el diagnóstico final por el neurólogo fue de síncope). Fueron derivados para valoración por cardiología y neurología infantil 14 (9,2%) y 11 (7,2%) niños respectivamente, siendo dados de alta con el diagnostico final de síncope vaso-vagal.
Conclusiones: El síncope vaso-vagal es una patología poco frecuente en la Urgencia de Pediatría. Una buena anamnesis y la ausencia de hallazgos patológicos en la exploración física hacen que en general la realización de pruebas complementarias y la valoración por otro especialista solo sea necesaria en casos seleccionados.
DIAGNÓSTICO POR IMAGEN Y TEMA LIBRE
P278 CARACTERÍSTICAS DE LA NEUROIMAGEN EN EL SÍNDROME DE SECKEL. 10:15 h
David Crespo Marcos, Amparo Carreño Beltrán, Aida de la Huerga, Jesús Cecilio López-Menchero Oliva, Lucía Múñoz, Adrián Ayala Garcés, Carmen Gutiérrez Regidor, Elena Cidoncha Escobar, Begoña Arias Novas, Dorotea Blanco Bravo
Hospital Materno Infantil Gregorio Marañón, Madrid.
Introducción: El síndrome de Seckel es síndrome raro de herencia autosómica recesiva. Cursa con retraso del crecimiento intrauterino, microcefalia, facies peculiar con prominencia de las estructuras del área central, retraso del desarrollo mental y enanismo armónico. Anteriormente sobrediagnosticado, se ha propuesto la inclusión de hallazgos característicos en la resonancia magnética cerebral.
Caso clínico: Recién nacido pretérmino (36+4s) de bajo peso (960g) para la edad gestacional fruto de un embarazo controlado con serologías negativas y sin otros factores de riesgo infecciosos. Amniocentesis 46 XY. En las ecografías presentaba una biometría de 16 semanas (en la 20) y de 27 en la 36. Cesárea por CIR, oligoamnios severo y flujo vascular fetal patológico. REA III, pH 7,31 y Apgar 7/9. Exploración inicial: Peso 960 g (< p3), Longitud 34 cm (< p3), P. Cefálico 26 cm (< p3). Fontanela amplia con suturas dehiscentes. Pabellones auriculares de implantación baja. Retrognatia leve. Labios finos. Microftalmía. Epicantus. Pies en mecedora. Evolución: tras estabilización inicial, comenzó con estridor inspiratorio y dificultad respiratoria, siendo diagnosticado tras fibrobroncoscopia de laringomalacia y estenosis subglótica circular congénita grado II-III de Cotton, que precisó corrección quirúrgica mediante Split cricotiroideo. Padeció 2 ITU por Klebsiella pneumoniae, objetivando tras CUMS reflujo vesicoureteral derecho grado II. Otros estudios: Cariotipo: 46 XY sin anomalías/ Ecocardiografía: FOP y estenosis pulmonar periférica/ Otoemisiones acústicas: normal/ Digestivo: intenso RGE espontáneo con cardias casi permanentemente abierto (tránsito gastro-intestinal), con pHmetrías normales/ Serie ósea: descarta displasia ósea/ Oftalmológico: retina y cristalino normal/ RM cerebral: retraso en patrón de girificación, con discreta plagiocefalia derecha. Microcefalia tanto supra como infratentorial generalizada/ Inmunoglobulinas séricas: IgG, IgA, IgM en límites normales/ Inmunidad celular: normal.
Conclusiones: Debe sospecharse un síndrome de Seckel ante un CIR armónico de causa desconocida con microcefalia, protusión de las estructuras mediofaciales y curva ponderoestatural retardada. En ausencia de otras malformaciones asociadas, una resonancia magnética con patrón de maldesarrollo cortical apoyará el diagnóstico.
P279 PREVALENCIA DE DIFERENTES HÁBITOS BUCALES EN NIÑOS VALENCIANOS. 10:20 h
Vanessa Paredes Gallardo, Bernardo Mir Plana, Carlos Paredes Cencillo
Centro de Salud Serrería I, Valencia, Hospital Clínico Universitario, Valencia y Facultad de Medicina, Valencia.
Introducción: Los hábitos orales son costumbres adquiridas por la repetición continuada de una serie de actos que sirven para calmar una necesidad emocional. Todos los hábitos anómalos modifican la posición de los dientes, la relación y la forma que guardan las arcadas dentarias entre sí en mayor o menor medida.
Objetivos: Los objetivos del presente trabajo fueron estudiar la prevalencia de los diferentes hábitos bucales en una población infantil de niños valencianos, así como observar las alteraciones producidas por lo mismos a nivel de la cavidad bucal.
Material y métodos: Por lo tanto, se estudió la presencia de estos hábitos en una muestra de 1100 niños valencianos entre los 6-11 años de edad que acudieron al un Centro de Salud de la ciudad de Valencia entre los meses de Enero a Junio de 2003. Los hábitos detectados fueron: el Bruxismo, la succión digital, la utilización del chupete, la deglución atípica y la respiración oral.
Resultados: Los resultados mostraron una distribución desigual en la presencia de los diferentes hábitos y parecida a los resultados registrados por otros autores sobre otras poblaciones. Las alteraciones encontradas a nivel de la cavidad bucal fueron las esperadas e iguales a las de otros investigadores sobre los mismos hábitos.
Conclusiones: Las conclusiones de nuestro estudio fueron: 1) Los hábitos anómalos se presentaron en la mitad de los niños valencianos estudiados. 2) Los hábitos anómalos se presentaron de igual manera en niños/as. 3) La deglución atípica fue el hábito que se apareció con mayor frecuencia en estas edades. 4) La mordida abierta fue la alteración bucal predominante. 5) El pediatra debe identificar estos hábitos bucales de manera precoz para poder ser tratados adecuadamente.
P280 TINCIÓN CROMÓGENA: UN PROBLEMA HABITUAL EN LA CLÍNICA PEDIÁTRICA. 10:25 h
Vanessa Paredes Gallardo, Bernardo Mir Plana, Carlos Paredes Cencillo
Centro de Salud Serrería I, Valencia, Hospital Clínico Universitario, Valencia y Facultad de Medicina, Valencia.
Introducción: La tinción cromógena supone un a alteración frecuente en la clínica pediátrica poco conocida y que preocupa a menudo a los Pediatras que la detectan. Esta tinción formada por unas manchas pequeñas y frecuentes de color negro, presenta una localización característica, ya que aparece pegada al borde más cercano a la encía del diente en cada paciente. La etiología de esta tinción es un tema controvertido, ya que cada autor, ofrece su opinión, muchos confirman su origen desconocido asociado posiblemente a la presencia de microorganismos en la saliva del paciente con sulfuro de hierro insoluble.
Objetivos: Los objetivos de nuestro estudio fueron: Calcular la prevalencia de la tinción cromógena en una muestra de niños valencianos estudiados y observar si la afectación era mayor en niñas que en niños.
Material y métodos: Por lo tanto, se estudió la presencia de esta tinción en una muestra de 1100 niños valencianos entre los 4-10 años de edad que acudieron al un Centro de Salud de la ciudad de Valencia entre los meses de Mayo a Octubre de 2003.
Resultados: Los resultados mostraron una distribución baja e igual en niños que en niñas de la tinción cromógena y muy similar a los resultados encontrados por otros autores. La tinción afectaba a todos los dientes de igual manera.
Conclusiones: Las conclusiones de nuestro estudio fueron: 1) La tinción cromógena se presenta con una prevalencia del 7,54%. 2) La tinción cromógena aparece de igual manera en niños que en niñas. 3) La tinción cromógena aparece de igual manera en dientes anteriores que posteriores, así como dentición temporal y permanente. El tratamiento de la tinción cromógena consiste únicamente en la realización de limpiezas mecánicas profesionales de los dientes periódicamente para eliminar la coloración de los mismos.
P281 TRANSTORNOS DEL CICLO DE LA UREA TARDÍOS: ¿FORMA DE PRESENTACIÓN MÁS FRECUENTE?. 10:30 h
Julia Balaguer Guill, Lara García Almiñana, Julia Apolinar Valiente, Jaime Dalmau Serra
Hospital Infantil La Fe, Valencia.
Introducción: La función del ciclo de la urea es eliminar el nitrógeno sobrante de la dieta. Clásicamente, los transtornos del ciclo de la urea se describen como de presentación neonatal, con convulsiones. De un total de 7 pacientes con transtornos del ciclo de la urea controlados en la Unidad, 4 de ellos fueron de presentación supuestamente atípica (tardía y sin convulsiones).
Casos clínicos:Caso 1: Niña de 16 meses que presentaba episodios de vómitos y decaimiento desde los 6 meses, que mejoraban con dieta. Por persistencia de episodios repetidos se solicita analítica en la que se objetiva GOT 70-100 UI/L. En el transcurso de su estudio, a los 15 meses ingresa por fallo hepático agudo, siendo la GOT > 300 UI/L. Caso 2: Niña de 4 años que ingresa en coma; diarrea y vómitos, obnubilación y dificultad de la deambulación. Al ingreso se palpaba hepatomegalia de 2 cm. GOT 529 y GPT 149 UI/L, tendencia a la alcalosis metabólica. Caso 3: Niña de 22 meses que ingresó por ataxia asociando transtorno de la conducta y vómitos. Durante el último año 1-2 episodios similares al mes, de 2 dias de duración, algunos coincidían con infecciones. Empeoramiento neurológico progresivo y coma. GOT 120 y GPT 103 UI/L. Caso 4: Niño de 15 meses, vómitos y somnolencia, 2 episodios anteriores similares. Deterioro neurológico y coma. GOT 68 y GPT 87 UI/L. Diagnóstico: Por hipertransaminasemia y clínica neurológica no filiada se hizo amonio (> 170 µM), confirmándose con aminoácidos del ciclo de la urea (glutamina > 1000 µM). Evolución favorable en todos, con restricción proteica, aminoácidos esenciales, arginina y quelantes de nitrógeno.
Discusión: Ante un clínica inespecífica neurológica, repetitiva, que mejora con dieta y/o hipertransaminasemia, tras descartar otras etiologías más frecuentes como infecciones de SNC, tumores intracraneales y causas tóxico-medicamentosas; se debe solicitar amonio y aminoácidos en la fase aguda. De 7 casos totales controlados, 4 fueron tardía (en > 6 meses); 3/4 con vómitos, somnolencia (3/4), coma (3/4), alteración de conducta (2/4) y ataxia (1/4). Hipertransaminasemia en todos. Ninguno presentó convulsiones. Clínica no fue desencadenada tras ingestas proteicas elevadas, dudosa relación con procesos infecciosos intercurrentes. Ante clínica digestiva y/o neurológica inespecífica y repetitiva e hipertransaminasemia no filiada, se sugiere estudio metabólico independientemente de la edad y de la falta de convulsiones.
P282 LA EXPERIENCIA DE LA TELEMEDICINA EN LA CIUDAD DE BUENOS AIRES. 10:35 h
Ariel Melamud, Julio Puiggari, Edgardo Checcacchi
Hospital General de Niños Pedro de Elizalde, Buenos Aires (Argentina).
Objetivos: El objetivo del presente trabajo ha sido analizar los resultados del proyecto de Telemedicina desarrollado en un Hospital Pediátrico desde 1998 y los motivos que dieron lugar a los resultados obtenidos.
Introducción: Interpretando que la Telemedicina es el elemento que reúne tecnología y comunicación el Gobierno de la Ciudad de Buenos Aires estableció la creación de la Red de Telemedicina de la Ciudad de Buenos Aires por disposición N° 149/DGAS/98 la cual inició sus actividades formalmente en marzo de 1998 con el fin de crear una eficiente comunicación en el sistema de salud capitalino.
Material y métodos: Este proyecto fue diseñado en tres etapas. La primera involucró 5 hospitales y se desarrollo por voluntad de los profesionales de los hospitales interesados. La segunda realizada en el año 2000 incluyó 6 hospitales más y se realizó con la ayuda del Gobierno de la Ciudad de Buenos Aires. La tercera etapa no ha sido aún desarrollada por motivos que se considerarán en la exposición del trabajo.
Resultados: La actividad hasta el momento ha estado basada fundamentalmente en videoconferencias de carácter políticas o docentes y ocasionalmente con instituciones del interior del país o del exterior. No se ha podido implementar una red asistencial que permita además de la docencia, la interconsulta médica o el intercambio de servicios. El programa en los últimos años ha sufrido las vicisitudes de cambios de gobiernos, de adecuaciones de presupuesto, de cambios de políticas comerciales de las empresas intervinientes generadas por la crisis económica, social y de salud que afecta a la Argentina y es por todos conocida.
Conclusiones: El Proyecto Red de Telemedicina de la Ciudad de Buenos Aires no ha logrado aún lo que se propuso originalmente Esperemos que en el futuro, las causas existentes que han impedido que este programa se desarrolle adecuadamente puedan superarse y que se establezcan objetivos alcanzables con pautas presupuestarias adecuadas, capacitación profesional y técnica.
P283 TELEMEDICINA: UN FUTURO PARA LA ATENCIÓN Y FORMACIÓN PEDIÁTRICA. 10:40 h
Juan Alonso Cozar Olmo, Mateo Jesús Díaz Torres, Fernando Sánchez García, Manuel Jesús Cuenca Burgos, M. del Carmen del Águila Grande, Andrés Segura Giménez
Hospital La Inmaculada del S.A.S., Almería.
Antecedentes y objetivos: Utilizar el sistema de videoconferencia de Telemedicina para garantizar el acceso de cualquier niño a nuestro Servicio de Pediatría, evitando desplazamientos en un área geográfica dispersa, así como mantener una formación continuada del pediatra extrahospitalario en su propio centro de salud.
Métodos: Equipo técnico de Telemedicina, con videoconferencia.Se realiza selección de patologías susceptibles de valoración (enuresis, cefaleas, asma, seguimiento de diabetes infantil..), así como temas de interés para la formación continuada del pediatra de zona (manejo del niño asmático, nuevas vacunas en la infancia, fiebre en el niño, manejo de leches especiales...)
Resultados:1) Alta precoz hospitalaria, 2) Disminución lista de espera en Consultas Externas, 3) Fomenta la colaboración interniveles, 4) Actualización pediátrica para los pediatras de zona en temas de interés mutuo, sin desplazarse de sus centros de salud.
Conclusiones: La Telemadicina, se presenta como una herramienta de futuro para la atención hospitalaria del niño en zonas de gran dispersión geográfica, como la zona Norte de Almería, con implicación de pediatra hospitalario y de atención primaria en el proceso del niño. Asimismo, es de gran utilidad, para la formación continuada de los pediatras que trabajan en áreas rurales
P284 INCIDENTALOMAS RETROPERITONEALES EN LA INFANCIA (A PROPÓSITO DE 2 CASOS CLÍNICOS). 10:45 h
Christian Garriga Braun, Roi Piñeiro Pérez, Carmen Martínez González, Celia Gil López, Esther Aleo Luján, Esther Vaquero Sosa, Julio García Casillas
Hospital Clínico San Carlos, Madrid.
Introducción: Se denomina incidentaloma a todo tumor detectado de forma casual, en ausencia de una sintomatología específica, durante una exploración radiológica que se practica por sospecha de cualquier otra patología. La frecuencia de los incidentalomas en la edad pediátrica no está claramente establecida, ya que no existen estudios aleatorizados al respecto, aunque con el uso de forma casi rutinaria de las técnicas de imagen, es previsible que el hallazgo de estas masas sea cada vez más frecuente.
A continuación se presentan 2 casos clínicos de nuestro servicio de pediatría en relación con este tema:
Caso 1: Paciente varón de 11 meses de edad con antecedente personal de displasia renal multiquística, nefrectomizado de riñón izquierdo a los 8 meses de vida, en el que durante un estudio ecográfico de control, se objetiva una masa suprarrenal derecha de características sólidas. Tras completar estudio diagnóstico finalmente es diagnosticado de neuroblastoma en estadio 1.
Caso 2: Paciente varón de 11 años de edad, sin antecedentes personales de interés, que en el postoperatorio de una apendicectomía presenta un cuadro febril, por lo que se realiza ecografía abdominal para descartar colecciones intraabdominales, evidenciándose una masa renal informada como quiste complejo (entre las posibilidades diagnósticas: quiste complicado, absceso renal, lesión tumoral: variante quística de tumor de Wilms). Tras realizar estudio diagnóstico no se puede descartar ninguna de las posibilidades por lo que se realiza acto quirúrgico con extirpación de la masa que anatomopatológicamente es informada como quiste benigno.
Los estudios revisados en la literatura pediátrica se basan esencialmente en incidentalomas de localización hipofisaria, pero no hemos encontrado trabajos extensos acerca de los de localización retroperitoneal.
P285 MUERTE PEDIÁTRICA NO NEONATAL: ASPECTOS ASISTENCIALES EN ENFERMOS CON PATOLOGÍA DE BASE. 10:50 h
José David Herrero Morin, M. Nuria Fernández González, Mónica García González, Gil Daniel Coto Cotallo
Hospital Universitario Central de Asturias, Oviedo (Asturias).
Introducción y objetivos: Los avances médicos y sociales contribuyen al descenso de mortalidad pediátrica. Actualmente ésta se relaciona frecuentemente con procesos crónicos y terminales. Se pretende describir aspectos epidemiológicos y asistenciales de la mortalidad pediátrica no neonatal en Asturias y analizar las diferencias existentes en la asistencia médica de los pacientes con patología crónica de base y sin ella.
Material y métodos: Se estudian los éxitus de pacientes menores de 16 años no neonatos entre los años 1999-2003, ambos inclusive, en los hospitales Cabueñes y Universitario Central de Asturias. Mediante revisión de historias se recogen datos epidemiológicos, clínicos y asistenciales. Se analizan las diferencias entre los grupos de pacientes con y sin patología de base mediante el test chi-cuadrado y exacto de Fisher, considerando significativos valores de p < 0,05.
Resultados: Se estudian 48 éxitus, con una edad media de 5 años y medio, predominio de lactantes (20 pacientes) y del sexo masculino (ratio mujer/varón 1/1,4). Las muertes son más frecuentes en el primer trimestre del año (35,4%), siendo la causa principal las infecciones (neumonías fundamentalmente). Tres cuartas partes de los fallecidos tienen alguna patología de base (habitualmente encefalopatías, seguido de neoplasias). El 45,8% de los pacientes están en una unidad de cuidados intensivos pediátricos al producirse la defunción. Se intentan maniobras de reanimación en menos de la mitad de los éxitus. En pacientes con patología de base es significativamente menos frecuente el ingreso en UCIP, el empleo de medicación inotrópica y ventilación mecánica, y las maniobras de reanimación. No existen diferencias en la realización de exploraciones analíticas previas al éxitus.
Conclusiones: La patología infecciosa es responsable de gran parte de la mortalidad pediátrica no neonatal, incidiendo con más frecuencia en enfermos crónicos. Muchos éxitus se producen como fase terminal de una patología grave o en enfermos con deterioro importante por una enfermedad de base progresiva, lo que lleva en ocasiones a una limitación del esfuerzo terapéutico.
P286 SÍNDROME DE LA CIMITARRA. 10:55 h
Arantza Vivanco López, Olaia Sardón Prado, Nere Arostegi Kareaga, Patricia Esparza Paz, Izaskun Miner Kanflanka, Eider Oñate Vergara, M. Ángeles Izquierdo Riezu, Agustín Nogues Pérez, Ángeles M. Ruiz Benito
Hospital Donostia, San Sebastián (Guipúzcoa).
Antecedentes: El síndrome de la cimitarra es un drenaje venoso pulmonar anómalo con hipoplasia pulmonar en el que parte o todo el sistema venoso pulmonar no drena en aurícula izquierda, sino en otro colector venoso. Se presentan 2 casos de síndrome de la cimitarra diagnosticados en los 12 últimos meses.
Observación clínica:
Caso 1: Niña de 10 meses de edad que ingresa por bronquitis. Antecedentes personales: 2 ingresos previos por ictericia neonatal y bronquiolitis VRS (-). Rx tórax de ambos ingresos compatibles con atelectasia de base pulmonar derecha. Rx tórax al ingreso: condensación parenquimatosa en pulmón derecho. Rx tórax control: Hemitórax derecho disminuido, mediastino desviado a la derecha. Vaso en parte inferior del pulmón derecho compatible con síndrome cimitarra. AngioRM: Drenaje venoso pulmonar derecho en vena cava inferior. No venas pulmonares derechas. Valoración cardiológica: CIA tipo OS.
Caso 2: Niño de 24 horas de vida, que ingresa por fiebre. EF: Tonos cardíacos audibles en hemitórax derecho. Taquipnea. Rx tórax: Pulmón derecho disminuido de tamaño y retracción del mediastino. Imagen compatible con síndrome de la cimitarra. AngioRM: Drenaje venoso pulmonar anómalo parcial derecho con presencia de 2 colectores superior e inferior que drenan en vena cava superior e inferior infradiafragmática. Discreta dilatación de la arteria pulmonar derecha. Valoración cardíaca: Ductus persistente, CIA tipo OS, hipertensión pulmonar.
Comentarios: El síndrome de la cimitarra generalmente es asintomático y el diagnóstico suele ser un hallazgo casual en una Rx tórax (pulmón derecho de menor volumen, desplazamiento mediastínico hacia la derecha e imagen densidad agua paracardíaca derecha). Las infecciones broncopulmonares recurrentes (caso 1) y la HTP (caso 2) son las complicaciones más frecuentes que presentan estos pacientes.
P287 ENFERMEDAD DE KAWASAKI: REVISIÓN DE NUESTRA CASUÍSTICA. 11:00 h
Ana M. Pastor Gómez, M. Luisa Herreros Fernández, Juana Barja Tur, Alfonso González Laguillo
Clínica Moncloa, Madrid.
Objetivos: Revisión de nuestra casuística de Enfermedad de Kawasaki (EK)
Material y métodos: Se revisan restrospectivamente12 pacientes diagnosticados de EK en nuestro centro. Enero 96-febrero 2004.
Resultados: La edad media fue de 3 años (8m-7años). Relación de sexos 1:1. Estancia media 8 días. La estación y mes más frecuente fueron invierno (50%) y Enero (25%).
Fiebre de más de 5 días la presentaron el 91%, rango 4-20 días. El exantema, afectación de labios y orofaringe y conjuntivitis (11/12) 91%, adenopatía laterocervical 58%, descamación periungueal 33%, edema de extremidades 16%. Artritis de rodilla en un caso y de caderas en otro. Ninguno presentó dolor abdominal, hidrops vesicular o manifestaciones neurológicas. Un caso recibió la vacuna triple vírica 72 h antes y otros 3 se acompañaron de vómitos, diarrea tos y otitis media. Un caso fue una EK incompleta y otro fue remitido de otro centro con el diagnóstico inicial de sospecha de mononucleosis. Complicaciones: destaca la ausencia de aneurismas coronarios. Hallazgos analíticos: Leucocitosis de > 15.000 (33%), anemia (41%), trombocitosis (66%) rango 253-666.000. La VSG y PCR: 75% y 66%. En el 33% se alteró la función hepática. Las serologías (CMV y EBV) y frotis faringeos fueron negativos 100%. El 91% (11/12) recibieron IGEV, el 66% a dosis de 2 gr/kg; en el 90% de los pacientes que recibieron este tratamiento la fiebre cedió antes de las 48 h.
Conclusiones:1) Ausencia de complicaciones cardiovasculares. 2) Las características epidemiológicas, clínicas y analíticas son similares a otras series españolas salvo la no diferencia entre sexos en nuestra serie, la baja incidencia de descamación periungeal (33% frente a 83-93%) y leucocitosis 33% frente 60-90%. 3) La importancia de la sospecha diagnóstica de EK, incluso en formas incompletas, para una terapéutica eficaz que evite complicaciones cardiovasculares.
P288 MODELO EUROPEO DE EXCELENCIA: APLICACIÓN EN EL ÁMBITO DE UN SERVICIO DE PEDIATRÍA HOSPITALARIO. 11:05 h
Eduardo González Pérez-Yarza, E. Zavala, M.A. del Riego, M.A. Izquierdo, J. Landa, C. Nuño, M.A. Pérez-Argüello, Ángel Rey Otero, T. Zurutuza
Hospital Donostia, San Sebastián (Guipúzcoa).
Antecedentes: Desde 1995, Osakidetza-Servicio vasco de salud impulsa la autoevaluación de sus centros por medio del Modelo Europeo de Excelencia (EFQM), a nivel de equipos directivos. Durante el último año recomienda la utilización de este modelo en los servicios y unidades de gestión clínicas
Objetivo: Reflexionar en equipo sobre las actividades y resultados del Servicio de Pediatría, según el EFQM, y detectar y priorizar áreas de mejora y establecer los planes de acción.
Método:a) Constitución de un equipo de trabajo formado por 4 facultativos (responsables de áreas médicas) y 4 enfermeras supervisoras de diferentes unidades clínicas. b) Realización de la autoevaluación utilizando la Guía de Autoevaluación EFQM para Unidades de Gestión Clínica, que contiene una aplicación informática diseñada al efecto. c) Priorización de la áreas de mejora, nombramiento de responsables y establecimiento del cronograma.
Resultados: La puntuación global obtenida en la autoevaluación fueron 247,63 puntos, distribuidos en:

Se obtuvieron 100 áreas de mejora de las cuales se han priorizado 15 planes de acción a desarrollar en 12 meses.
Conclusiones: El EFQM permite la orientación de las acividades del Servicio de Pediatría hacia la excelencia, proporcionando un referente de objetivos, facilitando la participación y ejercitando la dinámica de grupos.
P289 MASTOCITOSIS CUTÁNEO-SISTÉMICA DE PRESENTACIÓN NEONATAL. EVOLUCIÓN DE 4 AÑOS. 11:10 h
Julio Guerrero Vázquez, Julio Guerrero Fernández, Francisco Russo de la Torre, Perpetua de Paz Aparicio, Santos Olmedo Sanlaureano, Andrés Garcés Ramos
Hospital del S.A.S. Punta de Europa, Algeciras (Cádiz) y Hospital Materno Infantil La Paz, Madrid.
El término genérico de Mastocitosis incluye un conjunto de trastornos caracterizados por la sobreproliferación y acumulación de células mastocitarias en diferentes tejidos del organismo. Los signos clínicos varían dependiendo de la importancia cuantitativa de los acúmulos, de los órganos en que asientan y de los efectos de los mediadores químicos liberados por los mastocitos. La mayoría de las veces es la piel el tejido involucrado pero también pueden serlo el esqueleto, la médula ósea, el tracto gastrointestinal y el SNC. La presentación connatal es muy infrecuente y aun más el desarrollo precoz de extensas y severas afectaciones extracutáneas. Presentamos la evolución a lo largo de los 4 primeros años de vida de un caso excepcionalmente severo.
Caso clínico: Varón hijo único de padres jóvenes y sanos. Al nacimiento presenta intenso eritema universal, múltiples lesiones nodulares ampliamente distribuidas y, a las tres horas, llamativas lesiones bullosas. Confirmada histológicamente la naturaleza del proceso se establece tratamiento con hidroxicina, cimetidina, ketotifeno y medidas tópicas y profilácticas. La respuesta es insatisfactoria pues presenta episodios de shock histamínico y, a los 6 meses, hepatoesplenomegalia. Se asocia prednisona pero ha de retirarse al desarrollar hipertensión arterial sin haber llegado a controlar el proceso. Se aprecia retraso en el desarrollo psicomotor y se establece un síndrome caquéctico. Las investigaciones analíticas seriadas muestran un importante incremento de los niveles de histamina y sus metabolitos en sangre y orina así como los de triptasa sérica. Estudios de imagen: panvisceromegalia abdominal, llamativas lesiones óseas y cierto grado de atrofia cerebral. En la actualidad son excepcionales y leves los episodios de erupción bullosa pero mantiene, acentuadas, el resto de las alteraciones. Los padres han rechazado la punción medular para el estudio de la médula ósea.
Comentarios: Se discute la patogenia y analizan las modalidades clínicas de las Mastocitosis subrayándose la dificultad para el encaje nosológico de algunas formas de presentación; se ofrecen las posibilidades terapéuticas y las limitaciones en la infancia de algunas propuestas (interferones α, Cladribina, etc). Dado el fracaso de las utilizadas y lo tórpido de la evolución se considera que en el paciente descrito el pronóstico es malo
P290 TRATAMIENTO CON ALFA-L-IDURONIDASA EN UN NIÑO CON ENFERMEDAD DE HURLER. EXPERIENCIA TRAS UN AÑO DE TRATAMIENTO. 11:15 h
Raquel Rubio Rubio, Sonia Abió Albero, Trinidad Ureña Hornos, José Juan Calvo Vera, Ana I. Fernández Lorente, Antonia Chabas, Paz Ruiz-Echarri, Carmen Campos, Antonio Baldellou Vázquez
Hospital Materno Infantil Miguel Servet, Zaragoza y Instituto Bioquímica Clínica, Barcelona.
La enfermedad de Hurler está causada por un defecto del enzima α-l-iduronidasa, que provoca un acúmulo anormal de glicosaminoglicanos (GAGs) en los lisosomas celulares. El trasplante de médula ósea había sido hasta ahora la única opción terapéutica, pero el uso de la terapia enzimática sustitutiva está empezando a ofrecer resultados esperanzadores, a causa de la mejoría clínica y bioquímica que los pacientes parecen experimentar. Se presenta la evolución de un niño con enf. de Hurler tratado con *-l-iduronidasa desde enero del 2003 hasta la actualidad.
Paciente: Varón de 2a.y 8meses que consultó por fenotípo y cuadro clínico compatible con enf. Hurler a los 3meses. El diagnóstico de MPS I se realizó mediante hallazgo de patrón característico de GAGs en orina, análisis de actividad enzimática en fibroblastos y estudio genético mutacional.Tras ello, se inició la búsqueda de donante para TMO, no encontrándose ninguno compatible, por lo que en Enero 2003 se comenzó tratamiento con α-l-iduronidasa según protocolo habitual, con admon. semanal a dosis de 100ui/kg iv.
Resultados: La hepatoesplenomegalia disminuyó hasta alcanzar la normalidad, y se objetivó un descenso del 82% en la eliminación de GAGs en orina ya al 6º mes de terapia. Se observó mejoría progresiva respecto a las limitaciones en movilidad articular de rodillas, hombros y codos. A los 24 meses se consiguió la deambulación autónoma. Se ha objetivado también una menor incidencia de procesos infecciosos respiratorios. Sin embargo, no se han encontrado cambios en las opacidades corneales ni en el grado de hipoacusia. Tampoco se han demostrado por el momento cambios significativos en el desarrollo cognitivo del niño. En un año de tratamiento, no se ha producido ningún tipo de reacción adversa. Tras un año de tratamiento con α-l-iduronidasa, la mejoría clínica y analítica es evidente. A pesar de que persisten algunos problemas clínicos importantes y existen dudas acerca de la evolución a largo plazo, la patente mejora en la calidad de vida del paciente y la ausencia de reacciones adversas, justifican la terapia enzimática sustitutiva. Así, en el momento actual, y sin otra alternativa terapéutica, la administración del enzima parece un tratamiento apto y bastante seguro para aquellos pacientes afectos de MPS I que no puedan beneficiarse del TMO.
P291 ENFERMEDADES DE DECLARACIÓN OBLIGATORIA EN LA INFANCIA. 11:20 h
María Teresa León Espinosa de los Monteros, Ana Mª Pérez Aragón, Begoña López Hernández, Mª. Dolores Castillo Sánchez
Hospital Virgen de las Nieves, Granada.
Objetivos: Determinar la frecuencia de las enfermedades de declaración obligatoria (EDO) en niños de 0-14 años en Granada capital en el período 1998-2000. Conocer las enfermedades predominantes y su frecuencia por sexos.
Material y métodos: Estudio descriptivo de los datos suministrados a partir de la declaración de las enfermedades de declaración obligatoria (EDO) comunicados al Distrito de Granada capital durante el período enero 1998 a enero 2000. Se han excluido los casos de toxiinfección alimentaria. El análisis estadístico (descriptivo) se ha realizado con el programa SPSS.
Resultados: 354 casos han sido declarados durante el período de estudio, siendo 349 de los que se dispone de datos. 64,4% son varones frente a 34,7% de mujeres, siendo la enfermedad más frecuente en general la tuberculosis. Entre los niños de 0-14 años, (edad media 5,82 ± 3,5), las EDOs suponen el 19% (67) de las declaraciones. 53,7% son varones y 46,3% mujeres. La enfermedad más frecuente ha sido la parotiditis (32) 47,8%, seguida por la enfermedad meningocócica (11) 16,4%.

Conclusiones: Las EDOs suponen en la infancia el 19% del total de declaraciones, observándose un predominio en varones. Las enfermedades más frecuentes son las parotiditis, la enfermedad meningocócica y la tuberculosis, por orden de frecuencia.
P292 CASOS DE ERITEMA FIJO MEDICAMENTOSO EN LA INFANCIA. UN DIAGNÓSTICO NO SIEMPRE FÁCIL. 11:25 h
Jaime Lozano Blasco, Antonio Martínez Roig, Nuria López Segura, Verónica Seidel Padilla, Josefina Díaz Ledo
Hospital del Mar, Barcelona.
Introducción: Presentamos 4 casos diagnosticados durante los últimos 15 años en nuestro Servicio de Pediatría, con sus características clínicas, producidos por 3 fármacos distintos (cotrimoxazol, fenobarbital, y paracetamol).
Casos clínicos: No existían antecedentes de alergia familiar. Antecedentes patológicos a destacar en los casos 1, 3 y 4 eran las infecciones de vias respiratorias altas, mientras que el caso 2 era un niño con convulsiones febriles repetidas sujeto a tratamiento con fenobarbital. La edad de inicio oscilaba entre los 4 y los 13 años, y la localización era variable en todos ellos. Las caracteristicas de la lesión eran maculas violaceas (en los casos 1, 2, y 3) y/o marrones (en los casos 2, 3 y 4), y eritematosas (en el caso 3); con ampollas en el 3 caso.
Conclusiones: La erupción fija medicamentosa por fármacos es un proceso inflamatorio cutáneo, que se caracteriza por la recidiva de lesiones circunscritas a una o varias localizaciones cada vez que se administra un fármaco. El diagnóstico es fundamentalmente clínico; el tipo de lesión, la coloración, las localizaciones iniciales, y la evolución nos pueden orientar; es poco habitual hacer el diagnóstico en el primer episodio. Los fármacos con los que se relaciona suele ser analgésicos-antitérmicos-antiinflamatorios, sulfamidas, y antibioticos. En algunos casos algunos alimentos o aditivos, colorantes o sustancias preservativas. La edad media de presentación es aproximadamente 7 años. En la fase inicial la lesión consiste en una mácula eritematosa bien definida, redondeada u ovalada, de tamaño variable que con los dias adquiere una coloración rojo violácea. La mácula puede evolucionar a placa edematosa, o si la reacción es muy intensa llegar a formarse ampollas o vesículas en la zona central. Puede dejar una pigmentación residual de tonalidad marrón que puede durar semanas o meses. No es infrecuente la sensación de quemazón y prurito en las lesiones. Suele aparecer entre 30 minutos y 8 horas posteriores a la administración del desencadenante. La localización predilecta son las extremidades, cavidad oral y región circundante, area perigenital, genital y periorbitaira. El diagnóstico es clínico. Histológicamente se aprecia un infiltrado de celulas mononucleares cerca de la unión dermoepidérmica. El único tratamiento es la evitación del desencadenante.
P293 MÚLTIPLES QUISTES FIBROOSEOS SIMULANDO CLÍNICAMENTE UNA OSTEOMIELITIS CRÓNICA. 11:30 h
Patricia Calleja Cabeza, Virginia Carranza Parejo, Francisco Morales Horrillo, Antonio González-Meneses López, José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Antecedentes y objetivos: Presentamos un caso clínico de una niña de 10 años con quistes óseos dada su escasa frecuencia.
Material y método: Niña de10 años que consultó por febrícula, artralgias en rodilla izquierda, articulación esterno clavicular izquierda, muñeca y tobillo derecho de 3 semanas de evolución.Primo hermano con sintomatología similar.
Exploración: Buen estado general, bien hidratada y perfundida, normocoloreada, dolor y protrusión de articulación esterno clavicular izquierda. Dolor y ligera tumefacción a nivel de epífisis radial distal derecha, ligero dolor de tobillo derecho. Cardiorrespiratorio sin hallazgos patológicos. Aftas bucales. No signos meníngeos.
Pruebas complementarias: Hemograma, PCR, frotis de sangre periférica y proteinograma normales, VSG 52 mm. Estudio radiológico: Imagen compatible con quiste óseo en articulación esterno clavicular izquierda, signos de osteopenia en mano y muñeca derecha con hiperostosis de periostio e hipocalcificación de diáfisis. TAC esterno clavicular: A nivel de la unión esterno clavicular proximal de la clavícula, se aprecia una lesión lítica expansiva con pequeña trabeculaciones en su interior, sin poder precisar si se trata de matriz condroide o de una imagen ósea secundaria a la fragmentación ósea. No se identifican imágenes de partes blandas.Se observa buena separación de los tejidos de vecindad. Gammagrafía ósea: Focos de actividad osteogénica aumentada en extremo proximal de clavícula izquierda con pool vascular positivo y zona metadiafisaria distal de radio derecho con focos patológicos más dudosos en las otras dos localizaciones. Los hallazgos compatibles con osteomielitis. Biopsia de quiste esternoclavicular izquierdo; Lesión fibroósea benigna con infiltrado inflamatorio crónico.
Resultados: Tras la realización de la biopsia y, descartando malignidad, la paciente no precisó de tratamiento quirúrgico para la resolución de las lesiones, administrándole exclusivamente analgésicos y siendo revisada periódicamente.
Conclusiones: Debido a la atípica presentación del caso tuvo que realizarse un amplio diagnóstico diferencial, debiendo descartar, tanto tumores óseos, (encondroma, encondromatosis múltiple, granuloma eosinófilo, quiste óseo aneurismático y displasia fibrosa), como artritis crónica juvenil o infecciones óseas. Aunque tras la realización de las pruebas complementarias el diagnóstico más probable era infección ósea tipo osteomielitis crónica fue finalmente el estudio anatomopatológico el que pudo aportar el diagnóstico definitivo.
P294 TRATAMIENTO COGNITIVO-CONDUCTUAL EN ADMINISTRACIÓN INTRAMUSCULAR DE QUIMIOTERAPIA: CASO CLÍNICO. 11:35 h
Maitane Salas Arrambide, Olga Gabaldón Poc, José Luis Mayoral Miravete, Rafael Guerrero Pereda, Mila Arce Rodríguez, Imanol Amayra Caro
Hospital Donostia, San Sebastián (Guipúzcoa) y Universidad de Deusto, Bilbao (Vizcaya).
Introducción: El dolor y la ansiedad asociados al tratamiento médico en oncología pediátrica es uno de los aspectos de la enfermedad que más molesta a los niños oncológicos. Los niños con leucemia pueden padecer hasta 300 inyecciones a lo largo del tratamiento y seguimiento de la enfermedad, y en muchas ocasiones los síntomas de ansiedad y dolor asociados a estos procedimientos no reciben un tratamiento eficaz, resultando comprometida la adherencia al tratamiento médico.
Caso clínico: Niño de 6 años y 7 meses con Leucemia Aguda presenta un nivel de ansiedad y dolor extremos ante la administración intramuscular de metotrexato, requiriendo restricción física. Se propone aplicar un tratamiento psicológico cognitivo-conductual para el tratamiento de estos síntomas. Durante una inyección intramuscular en el Hospital de Día, se toma la línea base, obteniendo auto-informes del niño sobre la ansiedad anticipatoria y el dolor experimentados con el procedimiento médico utilizando la escala FACES Pain Rating Scale. En la siguiente visita, antes de la administración de quimioterapia intramuscular, se lleva a cabo la intervención psicológica de una hora de duración que consta de los siguientes componentes: ejercicios de respiración, distracción, refuerzo y ensayo conductual. En esta sesión están presentes la psicóloga, el niño y su madre. Tras la intervención psicológica, se administra la quimioterapia. La psicóloga guía al niño para que utilice los ejercicios aprendidos, y obtiene auto-informes del niño sobre la ansiedad anticipatoria y el dolor en 5 inyecciones que tienen lugar una vez a la semana.
Evolución: La ansiedad aticipatoria del niño disminuyó un 50% tras la primera intervención psicológica, y esta disminución se mantuvo durante las siguientes inyecciones. El dolor disminuyó en la segunda inyección tras la primera intervención psicológica, y fue disminuyendo paulatinamente a lo largo de los diferentes procedimientos médicos realizados, hasta llegar a disminuir un 80% respecto a la línea base una vez transcurrido un mes.
Conclusión: Las técnicas cognitivo-conductuales han demostrado ser eficaces en el tratamiento del dolor y la ansiedad asociados a procedimientos médicos dolorosos en Pediatría, y su utilización disminuiría el sufrimiento de niños y familias, además de facilitar la labor del personal sanitario y favorecer la adherencia al tratamiento.
P295 ECOGRAFÍA GINECOLÓGICA EN EL DIAGNÓSTICO TEMPRANO DEL SÍNDROME DE ROKITANSKY. 11:40 h
M. Teresa Urgel Gómez, Beatriz Tresaco Benedi, Gloria Bueno Lozano, María Jesús Barco Marcellán, Mercedes Sobreviela Laserrada, Rafael González de Agüero, Jesús Mª Garagorri Otero
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción: El síndrome de Rokitansky (1838) se caracteriza por la existencia de malformaciones diversas a nivel de aparato genital femenino con asociación variable de malformaciones renales y vertebrales. La presentación clínica más frecuente es la de una adolescente entre 15 y 18 años que consulta por amenorrea primaria, habiendo presentado un desarrollo puberal normal.
Caso clínico: Paciente de 14 años que consulta por ausencia de menarquia tras haber iniciado telarquia y pubarquia de forma progresiva en los últimos 2 años. Encuesta nutricional y situación pondoestatural normales. No existe patología crónica de interés. Primera hija de matrimonio no consanguíneo sano. Menarquias materna a los 12 años y de abuela materna a los 14 años. Talla genética 159 cm (P25-50). Exploración física: Peso 49 Kg (P25), talla 164 cm (P75-90). Genitales externos femeninos normales con himen permeable. Desarrollo correspondiente a un estadio 4 de Tanner. Con el objeto de descartar un retraso puberal simple o un hipogonadismo se realizan las siguientes pruebas: hemograma y bioquímica normales, test de LHRH: LH y FSH pulsátiles y con niveles basales normales, 17-βestradiol 48,4 pg/ml (rango 0-160), prolactina 14,8 ng/ml (rango 3-32), TSH 2,2 mU/mL (rango 0,3-4,9), DHEA 1.299 ng/ml, androstendiona 3,58 ng/ml. Edad ósea 13 años (Greulich y Pyle). Ecografía ginecológica: ovarios de 39,4 x 21 x 20 mm con folículos < 7 mm, útero rudimentario de 28 x 21 x 17,1 mm, mucosa endometrial de 3,28 mm. Resonancia magnética: canal vaginal atrófico con fórnices bien desarrollado, esbozo atrófico de cuerpo uterino y ovarios normales. Urografía de eliminación: normal. Radiografía de columna lumbosacra: falta de fusión de arcos posteriores a nivel S1. Cariotipo 46 XX. De acuerdo con la clínica y las pruebas de imagen se diagnostica de síndrome de Rokitansky.
Comentarios: Se pretende resaltar la importancia y utilidad de la ecografía ginecológica durante el período de la adolescencia y su buena correlación con los hallazgos de la resonancia magnética. En este caso, ha permitido el diagnóstico temprano (antes de los 15 años) de una malformación congénita la cual, de no haber sido diagnosticada, podría haber debutado en forma de abdomen agudo con la consiguiente repercusión clínica y psicológica para la paciente.
CIRUGIA
P317 CUERPOS EXTRAÑOS ATÍPICOS EN LA EDAD PEDIÁTRICA. 12:00 h
Eva Mª Fernández Calderón, J. Pando Pinto, Emilio Blesa Sánchez, J.I. Santamaría Osorio
Hospital Materno Infantil, Badajoz.
La ingestión o aspiración de cuerpos extraños (CE) es un accidente frecuente en la infancia y conlleva una alta morbimortalidad.
1º Caso clínico: Paciente de 12 años que en el estudio de coxalgia derecha, se objetiva en RX abdominal la existencia en hipocondrio derecho de un CE metálico. En endoscopia digestiva no se visualiza CE ni lesiones focales. La ecografía y TAC abdominal confirman la presencia de un objeto metálico lineal, tipo aguja, en el lóbulo hepático izquierdo, sin lesión de estructuras vasculares ni parenquimatosas. No existían antecedentes de intervenciones quirúrgicas previas ni de ingestión de CE, dolor abdominal o hemoptisis. Hipotéticamente sugerimos que el CE alcanzó esta localización tan atípica, intrahepática, tras la ingestión de una aguja que migró desde el tracto gastrointestinal y perforando directamente la pared intestinal, se enclavó a nivel de cara interna hepática y con los movimientos respiratorios ascendió hasta alcanzar su posición final. Al tratarse de un hallazgo casual en una paciente asintomática, sin que la posición del CE se haya modificado en controles sucesivos y según la bibliografía consultada, se optó por tratamiento conservador.
2º Caso clínico: Paciente de 12 años con dolor abdominal en hipocondrio derecho, fiebre y vómitos de 48 horas de evolución. En ecografía abdominal se objetiva una gran litiasis biliar; se realiza colecistectomía por laparoscopia. La paciente reingresa una semana después con cuadro de tos, febrícula y posterior dolor costal derecho. En la RX de tórax se halla una imagen de condensación en lóbulo inferior derecho con atelectasia. En una nueva anamnesis, la niña refiere, que mientras jugaba con una espiga de trigo en la boca, la ingirió de forma accidental, sin episodio claro de sofocación. Se realizan dos fibrobroncoscopias, sin que se visualice CE. Dada la buena evolución de la neumonía el paciente es dada de alta. Un mes después (casi dos meses después de la primera consulta), ingresa por tos y esputos hemoptoicos de una semana de evolución, pérdida de peso e inapetencia. En la RX de tórax se objetiva foco de condensación a nivel del lóbulo inferior derecho, así como engrosamiento pleural adyacente. En TAC torácico se identifica en lóbulo inferior derecho un pequeño infiltrado pulmonar, una zona de condensación, con broncocele y reacción pleural. Se realizó una segmentectomía del lóbulo inferior derecho.
P318 ANOMALÍAS UTERINAS. 12:05 h
Ana García González, M. Isabel López Conde, Alba Manjón Herrero, José Luis Fernández Iglesias, Esther Vázquez López, Ramón Morales Redondo
Complexo Hospitalario Xeral-Calde, Lugo.
El desarrollo anómalo de la cavidad uterina presenta una incidencia que oscila entre uno por ciento y uno por mil. La falta de fusión de los conductos paramesonéfricos (de Muller) en una región determinada o en toda su longitud explica los diferentes tipos de duplicación uterina. Clínicamente puede ser asintomática o dar lugar a una amplia gama de manifestaciones, como son: amenorrea primaria, menstruaciones irregulares, dismenorrea, masa pélvica, infecundidad, aborto.
Presentamos dos casos de la variante más intensa de malformación uterina: el útero doble (didelfo) diagnosticados en nuestro hospital en un intervalo de 8 meses.
Caso A. Niña 12 años de edad que acude a Urgencias por dolor en cadera dcha. de 2 meses de evolución. Diagnosticada de escoliosis, recibía tratamiento sintomático con diclofenaco desde hacía 4 días. El dolor se había acentuado en los últimos 2 días, coincidiendo con la menarquia, y se acompañaba de tenesmo y polaquiuria. En la exploración destacaba masa abdominal en FID. Al hacer tacto rectal se palpaba tumoración. Se solicita Ecografía y TAC abdominal, informados como colección en FID. El Servicio de Cirugía descarta apendicitis. Vista por ginecólogo se decide intervención quirúrgica con los siguientes hallazgos: hemiútero dcho. no comunicante con cérvix, con hematometra de 6-7 cm. y anexo dcho. normal.
Caso B. Niña de 12 años remitida por su Pediatra por presentar masa ovárica izq. de 8 x 7 cm objetivada en Eco abdominal, realizada por estreñimiento y dolor abdominal de 1 mes de evolución. Relataba 3 menstruaciones, la última hacía 24 días. En la exploración física presentaba abdomen blando, sin masas palpables, con dolor a la palpación en FII. Tacto rectal: tumoración sólida que abomba en fondo de saco de Douglas. Fue intervenida quirúgicamente, con el diagnóstico de malformación uterina (útero doble), con hemiútero izquierdo sin comunicación al exterior, y anexos normales.
P319 ABDOMEN AGUDO DE ETIOLOGÍA INFRECUENTE. 12:10 h
Raquel Perera Soler, Cristina León Quintana, Begoña Martínez Pineda, Norberto Hernández Siverio González, Roque Abián Montesdeoca Melián, Raúl Cabrera Rodríguez, Jerónimo Feo González, Javier Fernández Sarabia, Judith Mesa Fumero, Eduardo Domenech Martínez
Hospital Universitario de Canarias, La Laguna (Santa Cruz de Tenerife).
Introducción: El bazo ectópico es una entidad rara por si misma siendo en general asintomático, y es especialmente difícil pensar en el como causa de dolor abdominal en pediatría.
Caso clínico: niña de 10 años que desde hace 1 semana presenta dolor abdominal epigástrico intermitente e intenso que se irradia a hipocondrio derecho asociado a vómitos con ausencia de deposiciones, pérdida de apetito e interferencia con el sueño. En la exploración física destaca regular estado general, el abdomen es duro, doloroso con defensa muscular acentuada palpándose tumoración en hipogastrio. La analítica sanguínea muestra elevación de reactantes de fase aguda, leucocitosis (90% Neutrófilos), Hcto: 34,7%, Hb: 11,2 gr/dl y resto normal. En la radiografía de abdomen se observa imagen densa suprapúbica y asas distendidas. Ecografía de abdomen: masa sólida de 13x10 cm en hipogastrio con escasa captación doppler y ausencia de bazo en localización habitual.
Evolución: Con todo lo anterior se realiza nuevo control analítico y de imagen (TAC abdominal), a las 48 horas del ingreso, objetivandose descenso del hematocrito y de hemoglobina, aumento de la leucocitosis y reactantes de fase aguda, así como presencia de un bazo aumentado de tamaño de localización ectópica con escaso realce al medio de contraste y distensión de asas intestinales. La laparotomía realizada evidencia torsión de pedículo esplénico por lo que se realiza esplenectomía. La evolución clínica fue favorable.
Conclusión: En el caso del diagnóstico de un bazo ectópico asintomático no es preciso tomar ninguna medida pero si presenta dolor abdominal hay que sospechar su torsión.
P320 DIVERTÍCULO PARAURETERAL. PRESENTACIÓN DE UN CASO. 12:15 h
Susana Vidal Piedra, Mª Lourdes Jiménez Hernández, Idoia Martínez Repáraz, Beatriz Sangrador Martínez, Laura Buesa Casaus, M. Reyes Mazas Raba, Lucía Díaz de Entresotos Villazán, Mercedes Sánchez Rodríguez, M. Inmaculada Fernández Jiménez, Ernesto de Diego García
Hospital Universitario Marqués de Valdecilla, Santander (Cantabria).
Introducción: El divertículo paraureteral es una protrusión de la mucosa en la pared posterolateral de la vejiga, adyacente al orificio ureteral. Presenta escasa prevalencia, con mayor incidencia en varones. Habitualmente es unilateral. Entre las complicaciones posibles se encuentran el reflujo vesicoureteral, obstrucción ureteral y obstrucción uretral, alteración de la función renal, displasia renal, infección y litiasis.
Caso clínico: Varón de 6 años de edad, sin antecedentes familiares ni personales de interés, con fiebre de 24 horas de evolución acompañada de disuria, polaquiuria, hematuria al final de la micción y, en las últimas 6 horas, dolor lumbar. En la exploración física destaca dolor difuso a la palpación abdominal, más intenso en hipogastrio y en hemiabdomen izquierdo con puñopercusión renal izquierda positiva. Pruebas complementarias: hemograma con leucocitosis y desviación izquierda. Pruebas de imagen (ecografía abdominal, urografía, CUMS y MAG-3): marcada hipertrofia de la pared vesical sugestiva de cistitis hemorrágica con ureterohidronefrosis izquierda grado III y pseudodivertículo vesical paraureteral izquierdo (Hucht), con función renal izquierda conservada aunque con respuesta algo enlentecida al diurético. Se realiza diverticulectomía intravesical, siendo la evolución postoperatoria favorable. El estudio anatomopatológico confirma la presencia de pared de divertículo paraureteral con caracteres sugestivos de origen congénito.
Conclusiones: El divertículo paraureteral suele ser un hallazgo casual si bien puede acompañarse de cuadros de infección de vías urinarias e incluso masas abdominales. En nuestro caso debutó con una cistitis hemorrágica. La técnica diagnóstica de elección es el cistograma; el diagnóstico adecuado permite la corrección quirúrgica precoz, evitando el desarrollo futuro de las complicaciones asociadas.
P321 CIRUGÍA LAPAROSCÓPICA EN EL REFLUJO GASTROESOFÁGICO. 12:20 h
Francisco Chaves Pecero, Ana Isabel Jimenez Lorente, Salvador Morales Conde, Mercedes Granero Asencio, Filiberto Ramirez Guruchaga, Federico Argüelles Martín
Hospital Universitario Virgen Macarena, Sevilla.
Antecedentes: El reflujo gastroesofágico es una entidad relativamente frecuente en Pediatría donde el tratamiento médico-postural va a conllevar la resolución del proceso en un alto porcentaje de los casos. En aquellos pacientes donde el reflujo persiste por encima de los dos años a pesar del tratamiento o en aquellos donde determinados factores asociados, como los procesos neurológicos, van a impedir la desaparición del reflujo, la cirugía va a estar indicada para restablecer la competencia del esfínter cardial manteniendo una deglución normal, sin disfagia, así como la posibilidad de eruptar y vomitar en caso necesario. Con la incorporación de las técnicas de de Cirugía Laparoscópica en Junio de 2.002 a su arsenal terapéutico, el Servicio de Cirugía Pediátrica del Hospital Universitario Virgen Macarena de Sevilla inicia el tratamiento quirúrgico del reflujo gastroesofágico con esta técnica.
Método: Presentamos los resultados clínicos obtenidos en 24 pacientes con Reflujo Gastroesofágico a los que le hemos practicado un cierre de pilares y una funduplicatura tipo Nissen-Rossetti por vía laparoscópica. Analizamos edad, sexo, estudios preoperatorios practicados (gastroduodenal, pHmetria, manometria y endoscopia), complicaciones intra y postoperatorias, con especial atención a la disfagia postoperatoria, tiempos operatorios, analgesia, estancia media y resultados clínicos y pHmetricos.
Resultados: A todos los pacientes se ha practicado preoperatoriamente estudio gastroduodenal, pHmetria y esofagoscópica, mientras que la manometría solo se ha podido realizar en 14. No ha existido ninguna complicación grave en la serie. La disfagia postoperatoria ha estado presente en el 58% de los casos, aunque siempre desapareció antes del mes. El tiempo operatorio ha sido de 1 hora y 15 minutos, mientras que la estancia media ha oscilado entre dos y tres dias. En el 12.9% de los casos ha permanecido la clínica.
Conclusiones: La vía laparoscópica aporta beneficios al paciente inherentes a la técnica mientras mantiene los mismos resultados que la cirugía tradicional. Manteniendo un tiempo operatorio similar al de la cirugía tradicional, se acortan los tiempos de estancia hospitalaria, se aumenta el confort del paciente mientras que los resultados estéticos son excelentes.
P322 CASO CLÍNICO TORSIÓN DE EPIPLÓN. 12:25 h
José Antonio Peña Zarza, José Antonio Gil Sánchez, Ignacio Bregante Ucedo, Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca (Baleares).
Introducción: Torsión de epiplón es un cuadro que provoca un compromiso vascular de la zona con isquemia infarto asociado. Se presenta de forma primaria o secundaria asociada a procesos locales. Entidad muy poco frecuente a tener en cuenta en el diagnóstico diferencial de abdomen agudo.
Objetivo: Presentación de caso clínico
Caso clínico: varón de 13 años que acude al servicio de urgencias en varias ocasiones por abdominalgia de 4 días de evolución. El dolor se localizó inicialmente en epigastrio para focalizarse posteriormente en fosa ilíaca derecha. Dolor de intensidad inicialmente leve que fue en aumento con discreta exacerbación postpandrial, de ritmo intermitente con períodos de mejoría y sin irradiación. Presentó algún vomito aislado y perdida del apetito. Aspecto y ritmo deposicional normal. No clínica miccional. Afebril.
Exploración física: Destacaba un aceptable estado general y un sobrepeso. Abdomen blando y depresible sin masas ni megalias doloroso a la palpación profunda en fosa ilíaca derecha, blumberg dudoso positivo, peristaltismo presente. Puño percusión lumbar negativa. resto de exploración por aparatos anodina
Pruebas complementarias Hemograma: leucocitos 12.500 u/mcl (60% neutrofilos), resto normal. Tira reactiva orina: negativa, Pcr:2,1, bioquímica normal, Rx abdomen: anodina, Eco abdominal: algo de líquido libre en espacio subhepático de significado incierto, no se observan signos de apendicitis.
Evolución: Tras 12 horas en observación presentó un aumento de la abdominalgia, con un intenso dolor en la palpación en la fosa ilíaca derecha y una defensa abdominal. Con la sospecha de apendicitis se decidió la intervención quirúrgica, se observó una torsión de epiplón mayor derecho con una zona adyacente infartada.
Conclusiones:1) Entidad poco frecuente a tener en cuenta en el diagnóstico diferencial del abdomen agudo. 2) La clínica de una apendicitis de evolución tórpida sin signos de infección debe plantear el diagnóstico. 3) Las pruebas complementarias (rx simple, eco y tac) una vez descartada la apendicitis pueden guiar el diagnóstico.
P323 LA IMPORTANCIA DEL DIAGNÓSTICO CLÍNICO EN LA DETECCIÓN PRECOZ DEL FEOCROMOCITOMA. 12:30 h
Ana López Saiz, Carmen Benlloch Sánchez, Francisco J. Escriva Peña, Joaquín Donat Colomer, Juan Brines Solanes
Hospital Clínico Universitario, Valencia.
Objetivos: El feocromocitoma es el tumor responsable del 1% de los casos de hipertensión en niños. Su tasa de malignidad es del 10% y el pronóstico, como en todo tumor maligno, se beneficia del diagnóstico precoz, como se refleja en el caso clínico que exponemos a continuación.
Caso clínico: Niña de 7 años, sin antecedentes familiares relacionados, que debuta con cefaleas paroxísticas, ante las cuales su pediatra detecta hipertensión arterial severa que se manifiesta tanto al medirla en el brazo como en el muslo (lo cual descartaba coartación aórtica como causa de la hipertensión). Se practican exploraciones complementarias, entre las que destacan: ecografía, que evidencia masa suprarrenal derecha confirmada por resonancia nuclear magnética, sin evidencia de extensión local ni torácica, y con flujo vascular renal normal. Gammagrafía con meta-yodo-bencil-guanidina negativa. Gammagrafía ósea con tecnecio negativa. En las pruebas de laboratorio destaca la existencia de valores muy elevados de noradrenalina. Se inicia tratamiento antihipertensor con fenoxibenzamina, consiguiendo un buen control de la tensión arterial, tras lo cual se interviene, llevándose a cabo exéresis completa y aspirado de médula ósea. El informe anatomopatológico confirmó el diagnóstico de feocromocitoma, con ausencia de infiltración de médula ósea. Actualmente, al año de la intervención, se mantiene asintomática sin medicación.
Conclusiones: El cuadro sintomático de feocromocitoma incluye, además de la hipertensión, taquicardia, estreñimiento, aceleración del metabolismo y sangrado gastrointestinal ocasional, síntomas todos ellos inespecíficos. El control de la tensión arterial en la consulta del pediatra de atención primaria y la realización de una ecografía son maniobras sencillas que faiclitan el diagnóstico precoz de este tumor potencialmente maligno, con el consiguiente aumento de la supervivencia.
P324 ATRESIA DUODENAL COMPLETA ASOCIADA A MALROTACIÓN INTESTINAL Y VENA PORTA PREDUODENAL. CASO CLÍNICO. 12:35 h
José Antonio Montalvo García, Ana López Saiz, Leandro Pico Sirvent
Clínica Quirón, Valencia y Casa de Salud-Hospital Católico, Valencia.
Antecedentes y objetivos: La atresia duodenal asociada a la presencia de una vena porta preduodenal es poco frecuente. Presentamos aquí la descripción de un caso clínico y su solución quirúrgica
Métodos y resultados: Recién nacida de 38 semanas de gestación y sexo femenino nacida de parto espontáneo, con diagnóstico ecográfico prenatal de polihidramnios y sospecha de atresia intestinal. Las radiografías simples practicadas al nacimiento evidenciaron obstrucción a nivel de la segunda porción duodenal. Una vez estabilizada la paciente en la U.C.I. pediátrica se intervino de urgencia constatando la presencia de una atresia duodenal condicionada por la presencia de una vena porta preduodenal y asociada a una malrotación intestinal incompleta. La disposición pancreática era normal. Se practicó duodeno-duodenostomía latero-lateral conservando la disposición de la vena porta. Se revisó el resto del intestino sin hallar otras alteraciones. La paciente permaneció en la U.C.I pediátrica con sonda nasogástrica y nutrición parenteral 5 días tras los cuales se inició tolerancia oral sin problemas y fue dada de alta al 8º día del ingreso. El estudio radiológico fue normal. La niña se halla asintomática en la actualidad.
Conclusiones: La presencia de una vena porta preduodenal asociada a atresia duodenal en un recién nacido no afecto de síndrome de Down es una posibilidad poco frecuente pero a tener en cuenta en estos pacientes. La práctica de una duodeno-duodenostomía entre la primera y la cuarta porción duodenal ofrece en nuestra experiencia buenos resultados
P325 SOBREINFECCIÓN DE MALFORMACIÓN ADENOMATOSA QUÍSTICA PULMONAR TIPO II EN LACTANTE VARÓN DE 10 MESES. 12:40 h
Mirian Gonzalez Macías, Belén Joyanes Abences, Esther Vaquero Sosa, Julio García Casillas
Hospital Clínico San Carlos, Madrid.
Lactante varón de 10 meses diagnosticado intraútero de malformación adenomatosa quística pulmonar tipo II, asintomático desde el nacimiento, que acude a urgencias por cuadro de tos y fiebre de hasta 40ºC de una semana de evolución, sin dificultad respiratoria ni otra sintomatología acompañante. Presentaba buen estado general, pero ante la escasa respuesta al tratamiento domiciliario que había estado cumpliendo en su domicilio, y por el riesgo de infección pulmonar del paciente, se realizó radiografía de tórax, en la que se apreció un infiltrado neumónico compatible con infección de la lesión adenomatosa quística en lóbulo medio derecho. Se decide ingreso para instaurar tratamiento con antibioterapia intravenosa, y tras los estudios radiológicos pertinentes para delimitar la lesión y la infección, se intervino realizando lobectomía del lóbulo medio derecho y segmentomía del lóbulo superior derecho. Tras la cirugía, que cursó sin complicaciones, el paciente se encuentra asintomático.
Conclusión: El tratamiento quirúrgico reglado es para la mayoría de los autores de elección tras el diagnóstico de malforación adenomatosa quística, presentando estos pacientes una calidad de vida excelente en su mayoría. No obstante, el seguimiento y observación en pacientes asintomáticos es una alternativa, prestando especial cuidado a las posibles complicaciones infecciosas.
P326 BOCIO MULTINODULAR COMO LESIÓN PREMALIGNA. 12:45 h
María Isabel Gallardo Fernández, Jorge Martínez Pérez, José L. Alonso Calderón, Arantxa Ríos González, Ana Belén Jiménez Jiménez, Julián Lara Herguedas
Hospital del Niño Jesús, Madrid.
Introducción: La enfermedad multinodular de la glándula tiroides es infrecuente en niños y adolescentes por lo que recibe escasa atención; sin embargo, cuando aparece tras una primera neoplasia, con irradiación previa, debe considerarse como lesión premaligna
Material y métodos: Paciente diagnosticado de Sarcoma de Ewing en fémur derecho a los 7 años, tratado con quimioterapia y radioterapia local, que precisó para consolidación del tratamiento transplante autólogo de médula ósea, que a los 12,5 años presenta tumoración cervical derecha, dura a la palpación, sin signos inflamatorios, que se desplaza con los movimientos deglutorios, con función tiroidea normal y aumento de tiroglobulina. En Ecografía se observan ambos lóbulos agrandados, con imágenes de tipo multinodular y una región hipodensa en lóbulo derecho de 1,5 x 1 cm. En Gammagrafía se observa pérdida de estructura anatómica habitual con múltiples zonas con depósito escaso de trazador alternando con otras de depósito normal (bocio multinodular). La PAAF informa de patrón sugerente de tumor con patrón folicular. Se realiza tiroidectomía total en dos tiempos.
Resultados: El estudio anatomopatológico de la pieza revela adenomas foliculares múltiples sin signos de malignidad. No presentó complicaciones y se mantiene asintomático con tratamiento hormonal sustitutivo con levotiroxina sódica tras 6 años.
Conclusiones:a) La edad media de aparición en la literatura es de 12,8 años, probablemente en relación con la explosión glandular de la adolescencia, b) En todos los casos se aconseja un seguimiento estrecho del bocio multinodular ya que el riesgo de malignización es pequeño pero real, c) En pacientes con irradiación previa o tras neoplasia previa debe considerarse lesión premaligna y debe extirparse, d) La ecografía aparece como la exploración diagnóstica más importante en su seguimiento.
P327 PACIENTE CON ENFERMEDAD DE HIRSCHPRUNG QUE DEBUTA EN ETAPA NEONATAL CON CUADRO DE INVAGINACIÓN INTESTINAL. 12:50 h
Iván Somoza Argibay, Alberto Sánchez Abuín, Jorge Liras Muñoz, Roberto Méndez Gallart, Manuel Gómez Tellado, José Ríos Tallón, Ernesto Pais Piñeiro, Diego Vela Nieto
Hospital Juan Canalejo, A Coruña.
Introducción: La invaginación neonatal es una entidad rara que acontece en el 0,3% de todos los casos de intususcepción y en el 3% de las obstrucciones intestinales neonatales. La mayoría de las veces se diagnostica tardíamente y se presenta como una enterocolitis necrotizante. El 58% de las veces se encuentra asociada una etiología identificable, entre ellas se ha visto asociada a síndrome de bilis espesa y a síndrome de colon izquierdo pequeño. La relación entre invaginación neonatal y enfermedad de Hirschsprung no ha sido publicada previamente en la literatura.
Caso clínico: Paciente neonato a término de 3.140 gr, fruto de embarazo no controlado. Remitido a nuestro Centro a los dos días de vida por sospecha de obstrucción intestinal. Clínica de vómitos biliosos y distensión abdominal desde las 24 horas de vida, tras haber realizado deposiciones meconiales. En imágenes radiográficas se observa obstrucción intestinal probablemente a nivel ileal. Se realiza Enema con Gastrografín®, evidenciándose invaginación ileocecal que se desinvagina sin dificultad, comprobándose el paso de contraste a íleon. Tras su estabilización evoluciona favorablemente, precisando "nursing" para realizar deposición. Al 5º día de ingreso presenta empeoramiento clínico con sospecha de enterocolitis. Es intervenido quirúrgicamente realizándose colostomía de descarga y biopsias intestinales. Las biopsias intestinales demostraron ausencia total de células ganglionares a nivel de recto-sigma. Actualmente se encuentra pendiente de descenso rectal.
Discusión: Debe descartarse la presencia de invaginación intestinal mediante la realización de enemas de contraste y ultrasonidos en los pacientes neonatos con sospecha de obstrucción intestinal. Se ha postulado la diferencia de calibre entre el íleon y la válvula ileocecal como causa de la invaginación ileocecal. Esta teoría podría explicar la posible tendencia a la invaginación en los pacientes con dilatación patológica del colon, como en la enfermedad de Hirschsprung, el síndrome de bilis espesa o el síndrome de colon izquierdo pequeño.
P328 OTOPLASTIA POSTERIOR MÍNIMAMENTE INVASIVA PARA EL TRATAMIENTO DE LAS OREJAS PROCIDENTES. 12:55 h
Alberto Sánchez Abuín, Iván Somoza Argibay, Jorge Liras Muñoz, Roberto Méndez Gallart, Manuel Gómez Tellado, José Ríos Tallón, Ernesto Pais Piñeiro, Diego Vela Nieto
Hospital Juan Canalejo, A Coruña.
Objetivo: Presentar un simple y rápido método en formato de video para la corrección de las orejas procidentes (Helix valgus) mediante el modelaje anterior minimamente invasivo del cartílago auricular a nivel del antehelix y la plicatura de la concha por vía posterior.
Introducción: Desde la clásica descripción de Mustardé (1963) de la otoplastia para orejas procidentes, numerosas modificaciones han sido publicadas con similares resultados. Si bien no existen controversias acerca de la validez del abordaje auricular posterior para la realización de la otoplastia, el remodelaje del cartílago por esta vía se ha mostrado inadecuado en muchos casos debido a la aparición de recidivas. La cicatriz de una incisión anterior para abordar y plicar el antehélix es estéticamente inaceptable.
Material y métodos: Presentamos 41 otoplastias realizadas en 21 pacientes empleando el procedimiento de abordaje anterior minimamente invasivo. Este consiste en la realización de varias incisiones longitudinales a lo largo del antihelix empleando un bisturí de hoja lanceolada (de timpanotomía) de 2 mm de largo y con 12 cms de largo y 1 mm de diámetro entrando por vía anterior. Una vez realizadas las condrotomías lineales, el modelaje posterior logra una adecuada plicatura del antihelix que se fija con suturas no absorbibles por vía posterior. De la misma forma la concha se sutura a la fascia premastoidea. Se reseca un segmento de piel retroauricular de 0,5 cms de ancho y se cierra con monofilamento absorbible intradérmica. El seguimiento ha sido de 6 meses a 4 años (media 18 meses).
Resultados: No hemos observado complicaciones precoces tras la cirugía. La lesión puntiforme anterior era inapreciable a los 15 días de la intervención. En ningún paciente se produjeron cicatrices hipertróficas en la incisión posterior. La corrección de la protusión auricular se consideró estéticamente buena en todos los pacientes. En 1 caso se evidenció hipercorreción auricular. Un paciente presentó una asimetría llamativa en la corrección bilateral.
Conclusiones: Si bien todas las técnicas de corrección de las orejas procedentes deben ser evaluadas en un seguimiento a largo plazo, la consecución de un procedimiento que no produzca cicatrices anteriores y que permita modelar el cartílago por vía anterior es siempre prometedor. Son necesarios controles posteriores para evaluar la efectividad de este abordaje mínimamente invasivo.
P329 REFISTULIZACIÓN DE ATRESIA ESOFÁGICA CON FÍSTULA TRAQUEOESOFÁGICA DISTAL. 13:00 h
Purificación Aguilera Sánchez, María Rodríguez Martínez, Eduardo López Candel, Salvador Fernández Dozagarat, Manuel Martín González, Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
Introducción: La refistulización es la 3ª complicación más frecuente de la Anastomosis termino-terminal de la Atresia Esofágica y se trata de la más grave potencialmente. Aparece como consecuencia de la tensión creada en los cabos anastomóticos seguida del escape de secreciones, lo que da lugar a una respuesta inflamatoria local con dehiscencia de las suturas, dando lugar seguidamente a la refistulización. La sospecha clínica de este problema debe plantearse ante síntomas respiratorios recurrentes asociados con la alimentación del niño poco tiempo después de la reparación quirúrgica de la Atresia de Esófago.
Objetivo: Se presenta el caso de un lactante intervenido quirúrgicamente a los 3 días de vida por Atresia Esofágica tipo C en el que aparece cuadro consistente en crisis de atragantamiento con tos y cianosis coincidentes con las tomas.
Casuística: Lactante varón de 40 días de edad que acude al Servicio de Urgencias Pediátrico del Hospital por cuadro de tos y cianosis en relación con la toma del biberón que le ocurre desde los 15 días de vida, día en que fue dado de alta de este hospital tras ser intervenido de Atresia Esofágica tipo C. Las crisis se estaban haciendo cada vez más frecuentes y algunas se presentaban ya sin relación con la alimentación. A la exploración se objetivaba moderado tiraje subcostal y en la auscultación se apreciaban roncus aislados bilaterales. En ese momento el cuadro fue calificado como Bronquiolitis. A los 2 días volvió a Urgencias por no remisión ni mejoría de la clínica y se decidió el ingreso para completar estudio. El estudio de imagen reveló la presencia de una fístula traqueoesofágica "en H" a la altura de la anastomosis termino-terminal esofágica, ante lo cual se decide la reintervención quirúrgica con sutura de la fístula, aplicación de sello de fibrina e interposición de parche de pleura parietal.
Discusión y conclusiones: La infrecuencia de este problema junto con la clínica simulando un cuadro bronquial hicieron orientar el diagnóstico en principio hacia causas médicas pero siempre debemos tener en cuenta los antecedentes y la posibilidad de la relación del cuadro actual con ellos debido a la potencial gravedad del mismo. Las pruebas de imagen mostraron claramente la refistulización (no suele ser fácil visualizarla en estos casos) y el niño fue reintervenido sin demora, encontrándose actualmente en perfecto estado y asintomático.
P330 INVAGINACIÓN INTESTINAL RECURRENTE ASOCIADA A INFECCIÓN BACTERIANA E HIPERPLASIA NODULAR LINFOIDE DE ÍLEON. 13:05 h
Ana López, Cecilia Martínez Costa, Carmen Benlloch Sánchez, Ana Roche, M. José López, Agustín Molina, M. Rosa Alpera Lacruz, Juan Brines Solanes
Hospital Clínico Universitario, Valencia y Universidad de Valencia, Valencia.
Objetivo: Se presentan 3 casos de invaginación intestinal recurrente coincidentes con infección por Yersinia enterocolítica y uno con Campylobacter jejuni en los que se demostró hiperplasia nodular linfoide (HNL) de íleon. Dos casos requirieron cirugía a pesar de haber erradicado la enterobacteria.
Casuística:Caso 1. Lactante varón de 11 meses de edad que durante un episodio de gastroenteritis aguda (GEA) desarrolla invaginación ileocecal confirmada por ecografía y resuelta con neumoenema. Presenta 3 crisis similares a los 4, 7 y 14 días desinvaginándose de nuevo con neumoenema. En heces se aisló Yersinia enterocolítica (virus y parásitos negativos) y se trató con cotrimoxazol oral con controles posteriores, negativos. Tras nueva recidiva es intervenido evidenciándose importante HNL practicándose plicatura ileocecal. Comportamiento digestivo normal a 1,5 años de seguimiento. Caso 2. Niño con episodios recidivantes de invaginación a los 14, 15, 23 y 24 meses de edad, resueltos con neumoenema. En el último se aisló en las heces Campylobacter jejuni, (parásitos y virus, negativos). En ecografía y en examen gastrointestinal con contraste se puso en evidencia gran HNL de ileon. El tratamiento con eritromicina erradicó la bacteria, permaneciendo asintomático 6 meses tras los cuales vuelve a invaginarse (3 episodios en 48 horas) practicándose laparotomía y plicatura ileocecal. Lleva un año asintomático. Caso 3. Niño de 5 años de edad con antecedente de GEA el mes previo que desarrolla dolor abdominal y hematoquezia confirmándose invaginación por ecografía y solucionándose con neumoenema. En heces se aisló Yersinia enterocolitica que se trató con amoxicilina-clavulánico. En transito baritado se evidencia HNL de íleon. Permanece asintomático 10 meses.
Comentarios: Los 3 casos coinciden con infecciones bacterianas que probablemente indujeron hiperplasia linfoide aunque probablemente deben estar implicados otros factores ambientales no conocidos puesto que las infecciones bacterianas son frecuentes y raramente cursan con invaginación intestinal.
ENDOCRINOLOGÍA
P331 SÍNDROME POLIGLANDULAR AUTOINMUNE TIPO I. A PROPÓSITO DE UN CASO. 12:00 h
Cristina Marimón Blanch, Cristina Blasco Valero, Marian Albisu Aparicio, Merce Boronat Rom, Montserrat Carreras, Neus Potau Vilalta, Vicente García-Patos Briones, Lourdes Loidi Fernández de Trocóniz, Antonio Carrascosa Lezcano
Hospital Materno Infantil Vall d'Hebrón, Barcelona y Hospital Clínico Universitario, Santiago de Compostela (A Coruña).
Caso clínico: Niña de siete años con candidiasis mucocutánea, artralgias y alopecia.
Enfermedad actual: Presenta un cuadro de gastroenteritis asociado a tetania y calcemia de 4,4 mg/dl, fosforemia de 7 mg/dl y PTH de 3,5 pg/ml. Ante la presencia de candidiasis mucocutánea, hipoparatiroidismo (dos criterios mayores), alopecia, displasia ungueal y dental severa (criterios menores) se diagnostica de síndrome poliglandular autoinmune tipo I.
Evolución: Se inicia tratamiento con calcio endovenoso durante la crisis aguda y calcitriol 50 mcgr/ día y calcio oral como terapia de mantenimiento. Se objetiva secreción adecuada de cortisol con anticuerpos anticápsula suprarrenal positivos. Su estudio HLA fue: DRB1.03,15, DRB3 y DRB5. En el estudio genético molecular se demuestra mutación del gen "AIRE" (exón 6, mutación R257X en el alelo materno y C32.2FSx372 en el alelo paterno).
Conclusión: El síndrome poliglandular autoinmune tipo I es una entidad poco frecuente que engloba síntomas prevalentes en la población infantil cuya asociación debería conocerse.
P332 AUTOINMUNIDAD TIROIDEA EN NIÑOS CON DIABETES TIPO 1. 12:05 h
Purificación Aguilera Sánchez, Moisés Leyva Carmona, Emilio José García García, M. Ángeles Llamas Guisado, Francisco Javier Aguirre Rodríguez, Pedro Cortés Mora, Raúl Sánchez López, Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
Objetivos: Calcular la prevalencia de autoinmunidad tiroidea en niños con diabetes tipo 1. Estudiar las variables relacionadas con la presencia de esta autoinmunidad y su repercusión clínica.
Pacientes y métodos: Estudio transversal de los niños y adolescentes diagnosticados de diabetes tipo 1 antes de los 14 años que actualmente se siguen en nuestra consulta y se han realizado en el último año analítica que incluya función tiroidea y anticuerpos antitiroideos (antiperoxidasa y antitiroglobulina). Recopilación de variables clínicas y analíticas de la historia clínica.
Resultados: Se incluyen en el estudio 127 niños, 70 varones y 57 mujeres, de edad al debut entre 1,0 y 13,6 años y edad en el momento del estudio entre 2,33 y 16,75 años. 13 pacientes presentan anticuerpos antitiroideos positivos (10,2% del total). Los niños con esta autoinmunidad no se diferencian de los que no la presentan en cuanto a edad, edad al debut, forma de debut, presencia de celiaquía u otra enfermedad autoinmune (en el niño o en sus familiares de primer grado), evolución pondoestatural, evolución del control metabólico, requerimiento insulínico, aparición de luna de miel. La única variable relacionada con la positividad de la autoinmunidad tiroidea fue el sexo femenino (12/13, p < 0,001). De los 13 pacientes con anticuerpos antitiroideos positivos, 4 han desarrollado ya hipotiroidismo primario.
Conclusiones: La prevalencia de autoinmunidad tiroidea en niños diabéticos es del 10,2%. El sexo es la única variable relacionada, siendo su frecuencia mayor en mujeres. La autoinmunidad tiroidea no tiene repercusión sobre la evolución de la diabetes, pero sí un alto riesgo de aparición de hipotiroidismo.
P333 HIPERANDROGENISMOS OVÁRICOS. A PROPÓSITO DE SIETE CASOS. 12:10 h
Susana González de la Gándara, Alfonso Rodríguez Albarrán, Elisa Vázquez Peñas, Jesús Prieto Veiga, Valentín Salazar Alonso-Villalobos
Hospital Universitario, Salamanca.
Objetivos: Nuestro estudio pretende valorar la forma de presentación, sistemática diagnostica, evolución de la sintomatología clínica y su respuesta al tratamiento.
Métodos: Estudio retrospectivo de siete pacientes atendidas en el servicio de Endocrinología pediátrica. Se realizó valoración clínica, estudio de imagen mediante ecografía pélvica y suprarrenal, determinación hormonal basal y test de estimulación con Leuprolide (ProcrinR) y ACTH (SynacthenR) Además de test de frenado con dexametasona (Fortecortin).
Resultados: La edad media al diagnostico fue de 14 años y 7 meses. El test de Synacthen permitió el diagnóstico de una hiperplasia suprarrenal congénita no clásica (HSCNC) y una forma de hiperandrogenismo suprarrenal funcional (HSF); ambas niñas evolucionaron a un hiperandrogenismo ovárico funcional (HOF); otra niña presentó el antecedente de pubertad precoz. El HOF fue diagnosticado mediante técnicas de imagen, valoración basal hormonal y test de Procrin. En el 85% de los casos el hirsutismo y el acné fueron la forma de presentación. En el 28,5% de las pacientes se halló un hiperinsulinismo por resistencia a la insulina Se instauró tratamiento con acetato de ciproterona y etinilestradiol en seis casos y el tratamiento específico de la HSCNC y de la pubertad precoz.
Conclusiones: Cuando el hiperandrogenismo aparece en la infancia debe considerarse como diagnósticos más probables la pubarquia prematura por HSF o por HSCNC. En la etapa postmenárquica el diagnóstico mas frecuente es el de HOF con o sin ovario poliquístico. El antecedente de pubarquia prematura facilita la evolución hacia el HOF. La medicación específica debe utilizarse largo tiempo (ciclos de 18-24 meses) ya que los resultados favorables tardan en manifestarse. Tras la retirada pueden reaparecer los síntomas tras un período de remisión más o menos prolongado
P334 ESTUDIO PRELIMINAR DEL EFECTO DEL TRATAMIENTO CON HORMONA DE CRECIMIENTO EN PACIENTES CON BAJA TALLA FAMILIAR. 12:15 h
Carlos Martín de Vicente, Ruth García Romero, Segundo Rite Gracia, José Ignacio Labarta Aizpún, Esteban Mayayo Dehesa, Ángel Ferrández Longas
Hospital Materno Infantil Miguel Servet, Zaragoza.
Introducción: Los pacientes con baja talla familiar presentan una talla adulta baja habiéndose sugerido la existencia de una resistencia parcial a la hGH.
Objetivo: Evaluar el efecto de la hormona de crecimiento a dosis farmacológicas en pacientes prepuberales con baja talla familiar durante un año.
Métodos: 10 niños (6 varones y 4 mujeres) con edad media de 6,06 ± 2,28 años tratados durante un año con hGH líquida a 0,5 mg/Kg/semana y diagnosticados de baja talla familiar en base a antecedentes familiares, criterios auxológicos y secreción normal de GH (GH media en test de estimulación: 13,7 ± 6,9 ng/ml). Talla genética (TH):-1,79 ± 0,55 (varones: 163,7 ± 3,02; mujeres: 153,4 ± 4,17). RN: peso medio: -0,88 ± 1,29; longitud 0,98 ± 1,62. Se han analizado los siguientes parámetros: talla (T), peso (P), edad ósea (EO), velocidad de crecimiento (VC), pliegue tricipital (PT), pliegue subescapular (PSE), IGF-1, IGFBP-3, función tiroidea, glucemia, hemoglobina glicosilada (HbA1c), colesterol, LDL, HDL. Los resultados se expresan en SDS. Estudio estadístico: comparación de medias y estudio de correlaciones (p < 0,05*, p < 0,01**, ns: no significativo).
Resultados:

Conclusión: El tratamiento con rhGH, que ha sido bien tolerado, durante un año, ha producido un incremento significativo de talla sin producir una aceleración inadecuada de la edad ósea.
P335 PROLACTINOMA EN ADOLESCENTE ASOCIADO A DÉFICIT DE HORMONA DE CRECIMIENTO. 12:20 h
María Verísima Barajas Sánchez, Cristina Ruiz Serrano, Miriam Blanco Rodríguez, Ruth González Crisóstomo, Ana Leal Orozco, J. Ignacio Lara Capellán, Mercedes Bernacer Borja
Fundación Jiménez Díaz, Madrid.
Introducción: Los prolactinomas son tumores poco frecuentes en la infancia y adolescencia, pero durante esta etapa tienen mayor capacidad invasiva. El prolactinoma puede originar retraso o detención de la pubertad y más raramente pubertad precoz. La talla baja, por déficit de hormona de crecimiento (GH), es una de las formas de presentación de este tumor.
Caso clínico: Paciente de 16 años evaluado por presentar talla baja. Su desarrollo pondero estatural no llamó la atención de sus padres hasta que observaron una detención del crecimiento durante los últimos 3-4 años. No existen antecedentes de talla baja familiar ni el paciente había sufrido traumatismo craneal durante la infancia. En la exploración física, su talla era de 148,5 cm (P < 3, DS-3,3) y su peso 45Kg. (P < 3). El desarrollo puberal se encontraba en el estadío I-II de Tanner, con un volumen testicular de 6,7 ml y ausencia de vello axilar y púbico. No presentaba ginecomastia, galactorrea ni alteraciones visuales.
Datos complementarios: Edad ósea de 13,5 años. Estudio hormonal: FSH 2 mUI/ml, LH 1 mUI/ml, testosterona total < 25 ng/dl, GH basal < 1 ng/ml, IGF1 63 mg/ml y Prolactina (PRL) 3.089 ng/ml. La función tiroidea y la reserva de cortisol fueron normales. No se obtuvo respuesta de GH tras estímulo con insulina, glucagón ni propanolol. El estudio de GH integrada dio un pico máximo de 8,9ng/dl con una media de 1 ng/dl. Todos los test de evaluación de GH se realizaron tras primación con testosterona. La Resonancia magnética de hipófisis mostró un macroprolactinoma con extensión intra, infra y suprasellar.
Evolución: Se inició tratamiento con bromocriptina observándose: disminución progresiva, hasta su desaparición, de la masa tumoral, descenso de los niveles de PRL, aparición de caracteres sexuales secundarios y recuperación del crecimiento con una talla de 167cm. al finalizar el seguimiento del paciente.
Comentarios: Algunos pacientes con Prolactinoma presentan retraso puberal y talla baja por déficit de GH. El tratamiento con agonistas dopaminérgicos puede inducir la recuperación del eje hipotálamo hipofisario, por lo que no parece necesario asociar tratamiento con GH recombinante humana en todos los casos.
P336 ENFERMEDAD DE ADDISON Y TIROIDITIS LINFOCITARIA CRÓNICA. 12:25 h
María José González García, Mercedes Herranz Llorente, María del Carmen Torres Torres, Francisco Javier Arroyo Díez, Valentín Carretero Díaz
Hospital San Pedro de Alcántara, Cáceres.
Introducción: La enfermedad de Addison (EA), insuficiencia suprarrenal primaria que se debe a la destrucción global y progresiva del córtex suprarrenal, origina deficiencia en la producción de glucocorticoides y en ocasiones de mineralocorticoides. La causa más frecuente de EA es la Adrenalitis Autoinmune, seguida de la etiología infecciosa (tuberculosis, micosis e histoplasmosis). En niños, la EA es rara, pero no excepcional. Las manifestaciones aparecen cuando hay una destrucción de 3/4 partes de la glándula suprarrenal, las más llamativas son hiperpigmentación, trastornos gastrointestinales y debilidad generalizada progresiva.
Caso clínico: Niña de 11 años remitida por pérdida de peso (10 Kg en 6 meses), hiperpigmentación cutánea y astenia. Diagnosticada de tiroiditis linfocitaria crónica (TLC) hipotiroidea (T4 libre 0,97 ng/dl, TSH 6,26 mcU/ml, anti-TPO 2.29 UI/ml), en tratamiento con Levotiroxina 25 mcg/día. Tensión arterial 83/38 mmHg. En analítica al ingreso: ACTH 5269 pg/ml (9-52), cortisol 0,7 mcg/ml (7-26), Na 130 mmol/l, Ac anti-corteza suprarrenal positivo 1/40. Se inicia trataminto con hidrocortisona (15-20 mg/m2/día, en 3 dosis), con rápida ganancia de peso y desaparición de sintomatología. Actulamente sigue controles periódicos vigilando una posible aparición de Diabetes Mellitus
Comentarios: En todo paciente con hiperpigmentación, astenia, pérdida de peso y alteraciones iónicas pensar en la EA como posible etiología. Ante una EA hay que descartar otros procesos autoinmunes y Adrenoleucodistrofia. Es necesario el inicio precoz del tratamiento para evitar posibles crisis suprarrenales agudas. Importancia de control periódico en pacientes con EA y TLC por la posibilidad de desarrollo de Diabetes Mellitus, constituyendo así un Síndrome Poliglandular Autoinmune Tipo 2.
P337 HIPERTIROIDISMO NEOTATAL. 12:30 h
María José García Arias, Almudena del Pino de la Fuente, Celia Gómez Robles, Juan Pedro López Siguero, María José Martínez Aedo Ollero, Antonio Jurado Ortiz
Hospital Materno Infantil Carlos Haya, Málaga.
Introducción: El hipertiroidismo neonatal está producido por el paso trasplacentario de anticuerpos estimulantes del tioroides (TSAb), siendo la causa más frecuente la enfernmedad de Graves materna. Presentamos el caso clínico de un RN con hipertiroidismo neonatal hijo de madre hipotiroidea por enfermedad autoinmune.
Caso clínico: RN mujer de 7 días de vida remitida desde la unidad de screening de hipotiroidismo congénito por presentar datos analíticos de hiperfunción tiroidea. Exploración física: BEG. FC: 140 lpm CC: NC. Fontanela normotensa. Tórax: NC. No distrés. ACP: No soplos. Abdomen: normal. GU: femenino normal. SN: activa y reactiva. Reflejos arcaicos presentes. Pruebas complementarias: TSH: 0,00 mcU/ml; T4- libre: 113.8 pmol/l; T3-libre: 19,61 pmol/l; TPO: negativo; Ac antireceptor de TSH: 60,2 UI/ml
Evolución: Se inició tratamiento con Lugol al 5% 3 gotas al día, Propanolol a 1 mg/Kg/día y Neotomizol 0,5 mg/ Kg/día. Permaneció ingresada con control de constantes durante 21 días. Al alta presentaba en el estudio tiroideo: TSH: 0,00 mcU/ml; T4- libre: 9,6 pmol/l; T3-libre: 4.7 pmol/l; Ac antireceptor de TSH: 6,3 UI/ml. Se mantuvo el tratamiento con Neotomizol, retirándose el tratamiento con Lugol y Propanolol. A la edad de 1.11/30 meses se retira Neotomizol.Desde entonces la paciente presenta valores de TSH y T4 libre normales, con persistencia de Ac antireceptor de TSH, que se negativizaron a la edad de 7 meses.
Conclusiones:1) La causa más frecuente de hipertiroidismo neonatal es la enfermedad de Graves materna. 2) Se recomienda que las mujeres embarazadas eutiroideas con antecedentes de enfermedad tiroidea autoinmune se sometan a la determinación de Ac antitiroideos alrededor de la semana 26 de gestación. 3) La enfermedad en el RN puede manifestarse en el naciemiento o con un intervalo variable de 10 días a 4-6 semanas, con una duración de 8 a 20 semanas. 4) El hipertirodismo neonatal se trata con fármacos antitiroideos y betabloqueantes sumados a la solución de Lugol.
P338 OBSERVACIONES DE TUMORES DE QUÍSTICOS OVÁRICOS EN LOS QUE ESTUVO INDICADO UN PROCEDIMIENTO QUIRÚRGICO. 12:35 h
Celia M. Rodríguez Rodríguez, Clara García Cendón, M. Carmen García Barreiro, Cristina Lorenzo Legeren, Susana Rey García, Patricia Pernas Gómez, Gemma Novoa Gómez, Federico Martinón Sánchez
Complejo Hospitalario, Ourense.
Introducción y justificación: Los tumores quísticos ováricos prepuberales son relativamente raros en las niñas y mayoritariamente benignos, incrementándose su frecuencia con la edad. En la pubertad continúan siendo predominantemente benignos; si bien, la mayoría son funcionales y con un índice de malignidad inversamente proporcional a la edad. Su diagnóstico y tratamiento en niñas y adolescentes es problemático y controvertido. Al aportar una casuística en que se requirió un procedimiento quirúrgico contribuimos al establecimiento de criterios uniformes para esa indicación.
Material y métodos: Estudio retrospectivo de 17 niñas de 36 semanas de edad gestacional a 14 años con tumores quísticos ováricos que por su sintomatología aguda o sospecha diagnóstica de posible malignidad fueron sometidos a tratamiento quirúrgico.
Resultados: De los 17 casos 2 se diagnosticaron durante el período fetal (11%) y los 15 restantes (88%) a la edad media de 11 años y 8 meses con un rango de 2 meses a 14 años y 8 meses. La expresión clínica predominante fue el abdomen agudo (11casos que corresponden al 64.7%), y las observaciones restantes fueron 6 casos, que corresponden al 35.3% con hallazgos radiológicos compatibles con malignidad. El diagnóstico anatomopatológico fue: quistes de paraovario 3 casos, entre los cuales 2 presentaban infarto hemorrágico, quiste ovárico torsionado 2 casos, cuerpo lúteo hemorrágico quistificado 8 casos, cistoadenoma mucinoso 1 caso y cistoadenoma seroso 1 caso.
Análisis y discusión de resultados: Nuestra serie confirma que el diagnóstico clínico y por métodos de imagen cualificados es difícil de establecer de manera indudable, dado que los hallazgos a veces son de transición y no permiten una indicación clara, como de hecho confirman el 47% de los casos, en los cuales el examen histomorfológico evidenció cuerpo lúteo hemorrágico; y por tanto, la actitud podría haber sido expectante o con supresión mediante tratamiento hormonal y seguimiento ecográfico de control, a pesar de su expresión clínica aguda justificante de la intervención quirúrgica.
P339 PATOLOGÍA MAMARIA EN LA NIÑA Y LA ADOLESCENTE: ANOMALÍAS CONGÉNITAS Y FALSOS TUMORES. 12:40 h
Susana Rey García, Gemma Novoa Gómez, Cristina Lorenzo Legeren, Patricia Pernas Gómez, M. Carmen García Barreiro, Celia M. Rodríguez Rodríguez, Clara García Cendón, Federico Martinón Sánchez
Complejo Hospitalario, Ourense.
Introducción: La patología mamaria corresponde a un capítulo de gran importancia dentro de la ginecología pediátrica, que despierta el interés de un número cada vez más significativo de pediatras, debido al incremento de la demanda asistencial por este tipo de trastornos, motivado probablemente por las campañas de prevención. Presentamos un estudio descriptivo de los trastornos mamarios de la niña y la adolescente en nuestro medio que contribuirá a delimitar las características de estos procesos y nos facilitarán un abordaje diagnóstico y terapéutico de los mismos.
Material y métodos: Comprende un estudio descriptivo longitudinal retrospectivo, en el que se recogieron 632 observaciones de trastornos mamarios de la niña y la adolescente. A cada una de las observaciones se les abrió un protocolo unitario de estudio en la consulta de Endocrinología Pediátrica, que incluyó: los datos anamnésicos, clínicos y de las exploraciones complementarias realizadas a cada una de las pacientes.
Resultados: Las anomalías congénitas representaron el 15% de los casos, y en su mayoría son malformaciones menores. Diferentes procesos mamarios han sido interpretados como tumores por las pacientes o sus familiares, con una frecuencia bastante elevada (19%); en contraposición con la patología tumoral que representa un 6%, la mayoría benignos (5,7%), constituyendo la patología quística (3%) y el fibroadenoma (1,42%) las entidades más frecuentes; frente a los tumores malignos que tienen un carácter excepcional. Otro motivo de consulta por la ansiedad que genera es la secreción por el pezón (4,90%).
Conclusiones: Las anomalías congénitas son en su mayoría malformaciones menores como la politelia o el pezón invertido, y tienen carácter excepcional la atelia, la amastia el pezón deprimido y el pezón bífido. Entre los procesos mamarios interpretados como tumores, destacan por su frecuencia: la mastodinia (6,17%) y la telarquia asimétrica (5,22%). La enfermedad mamaria benigna es una entidad poco definida que incluye incluso variaciones de lo normal, y que aparece como la forma más frecuente interpretada como tumor, seguida del verdadero tumor benigno más repetido, el fibroadenoma.
P340 DIABETES TIPO 2 EN POBLACIÓN PEDIÁTRICA. 12:45 h
Esmeralda Colino Alcol, Emma Lara Orejas, Marta López Capapé, Herminia Rodríguez, Margarita Revenga Parra, M. del Milagro Alonso Blanco, Raquel Barrio Castellanos
Hospital Ramón y Cajal, Madrid.
Durante la última década ha habido un incremento llamativo en la incidencia de Diabetes Mellitus tipo 2 (DM2) en la edad pediátrica en ciertos países. La incidencia varía con el grado de obesidad, la edad y etnia. La DM2 es un trastorno metabólico complejo de etiología heterogénea. En el desarrollo de la enfermedad intervienen factores genéticos y ambientales. No hay datos de prevalencia de DM2 en población pediátrica en España, en nuestra experiencia de 300 niños diabéticos solo 1% tienen DM2. Presentamos estos 3 pacientes.

Conclusiones:1) La DM2 es infrecuente en nuestra población pediátrica. 2) La DM2 en esta edad no siempre está ligada a la obesidad. 3) En los obesos suelen presentar datos de síndrome metabólico. 4) Aunque inicialmente puede controlarse con dieta y ejercicio, alguno precisa tratamiento con antidiabéticos orales.
P341 PREVALENCIA DE SOBREPESO Y OBESIDAD EN LA POBLACIÓN ESCOLAR ALMERIENSE. 12:50 h
Encarnación López Ruzafa, Moisés Leyva Carmona, Emilio José García García, M. Ángeles Llamas Guisado, Francisco Javier Aguirre Rodríguez, Pedro Cortés Mora, Raúl Sánchez Pérez, Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
Introducción: En la actualidad tanto el sobrepeso como la obesidad representan uno de los problemas de salud pública más importantes en los países industrializados. La obesidad infantil es un trastorno muy frecuente y de prevalencia creciente que repercute, no sólo en la salud del niño (morbilidades asociadas como enfermedades cardiovascular y diabetes, y su evolución natural hacia la obesidad del adulto) sino también en su adaptación social y desarrollo psicológico.
Objetivo: Estimar la prevalencia de sobrepeso y obesidad en la población escolar de Almería capital según el índice de masa corporal.
Material y método: Estudio observacional de tipo transversal en el que se incluyen 499 niños (245 niños y 254 niñas) con edades comprendidas entre los 7 y 14 años, que fueron elegidos de forma aleatoria entre la población escolar almeriense. Se determinó peso, talla e IMC (Peso/Talla2) y se compararon con las tablas de IMC validadas tras el estudio Enkid y con las de la International Obesity Task Force (IOTF).Se considerós sobrepeso los IMC comprendidos entre P85 y P97 y obesidad los situados por encima de P97.
Resultados: La prevalencia de sobrepeso en la población escolar de la capital de Almería se estimó en un 16,03% (50% niños y 50% niñas) y la de obesidad en un 10,22% (43.14% niños y 58,86% niñas).
Conclusiones: Tanto el sobrepeso como la obesidad infantil tienen una prevalencia muy elevada en nuestra capital. Comparados con estándares nacionales e internacionales nuestros niños tienen mayor sobrepeso y obesidad. Sigue siendo importante recomendar la inclusión de evaluaciones rutinarias de peso y talla además de programas de actividad física y alimentación bien estructurados.
P342 EVOLUCIÓN DEL PESO Y DEL ÍNDICE DE MASA CORPORAL EN NIÑOS OBESOS QUE ASISTEN A UNA CONSULTA DE ENDOCRINOLOGÍA PEDIÁTRICA. 12:55 h
Bartolomé Bonet Serra, M. Mercedes Bueno Campaña, Javier Pérez-Lescure Picarzo, Javier Ramírez Martín, Isabel Sánchez Vera, Marta Viana Arribas, Amalia Quintanar Rioja
Fundación Hospital Alcorcón, Madrid y Universidad San Pablo - CEU, Alcorcón (Madrid).
La obesidad es uno de los principales problemas sanitarios en los países occidentales, sin un tratamiento eficaz. En adultos se ha puesto de manifiesto que determinadas épocas del año son más propensas a la ganancia de peso.
Hipótesis: En niños obesos la ganancia de peso tiene lugar en los meses de verano y en la Navidad.
Objetivo: Determinar en niños obesos la ganancia de peso a lo largo del año. Estudiar si la evolución es diferente entre niños y niñas.
Diseño experimental: Estudio retrospectivo llevado a cabo en 42 niños y 45 niñas, con una media de edad respectivamente de 10,4 ± 0,7 y 11,8 ± 0,9 años que acuden a la consulta con el diagnóstico de sobrepeso y son seguidos en la misma con revisiones periódicas cada 3 meses. En todos ellos se realiza una encuesta dietética, ajustando su dieta a las recomendaciones correspondientes a su edad. También se indica la realización de deporte de forma regular. Para determinar la ganancia de peso a lo largo del estudio se estimo la variación en la Z-score del índice de masa corporal (IMC) y del peso en 4 períodos consecutivos del año que abarcan: la primavera, verano, otoño, invierno (incluyendo Navidad). El seguimiento se realizo durante un año y medio, en los años 2002 y 2003. La comparación entre los grupos se llevó a cabo mediante el test de la t de student para muestras pareadas (*p < 0,05). *: muestra las diferencias entre un período respecto del anterior. Los resultados se expresan como media ± error estándar.
Resultados:

Conclusiones: En los niños obesos, durante el tiempo de seguimiento se observa una estabilización del peso, con una disminución del mismo durante el curso escolar. En las niñas el peso no varía a lo largo del estudio. Estos datos sugieren que el tratamiento de la obesidad debe ser la prevención de la misma, dada la dificultad de perder peso, sobre todo en el sexo femenino.
P343 PUBERTAD PRECOZ CENTRAL ASOCIADA A NEUROFIBROMATOSIS TIPO I. 13:00 h
Lorea Ruiz Pérez, María Zapico Álvarez-Cascos, Cristina Moscardo Guilleme, Carlos T. Esquembre Menor, José Flores Serrano
Hospital General Universitario, Alicante.
Introducción: La neurofibromatosis tipo I (NFI) es el trastorno neurocutáneo más frecuente y una parte de los mismos desarrolla pubertad precoz central (PPC). Las causas principales son la existencia de gliomas ópticos, la necesidad de radioterapia intracraneal, así como factores de crecimiento y neurotransmisores que aparecen en esta enfermedad. Presentamos el caso de una niña diagnosticada de NF I que desarrolló en su evolución una PPC.
Caso clínico: Niña de 3 años, diagnosticada de NFI con glioma de quiasma y nervio óptico bilateral, que fue tratada con quimioterapia citorreductora y posterior abordaje quirúrgico. Antecedentes familiares: madre y hermano afectos de NFI. Acude a consulta por telaquia precoz. Exploración física: Múltiples manchas de café con leche. Escaso panículo adiposo. Estadio de Tanner (telarquia [S4], pubarquia [P1]), no axilarquia. Exploraciones complementarias: Edad ósea mayor en un año a la cronológica. Test estímulo con LH-RH y estradiol: niveles puberales. Resto estudio hormonal normal. Ecografía abdominal: útero de morfología y tamaño adulto. Ante el diagnostico de PPC se decidió iniciar el tto con análogos de la LH-RH a los 4 años de edad cronológica, con enlentecimiento de la velocidad de crecimiento y detección de desarrollo de caracteres sexuales secundarios. A los 5 años presenta un desarrollo mamario S3, pubarquia 1 y maduración ósea de 6 años. Test de estímulo con LH-RH y estradiol: niveles infantiles.
Conclusión: Existe un relación entre la NFI y el desarrollo posterior de PPC, por tanto, es muy importante el seguimiento de estos niños para realizar un diagnóstico temprano de la pubertad precoz e instaurar rápidamente el tratamiento con análogos de LH-RH. Así, podremos detener la maduración gonadal, el avance de los caracteres sexuales y la maduración ósea, para alcanzar una talla final adecuada.
P344 INDICADORES DE ESTRÉS OXIDATIVO EN NIÑOS CON BAJA TALLA TRAS TEST DE ESTÍMULO CON HIPOGLUCEMIA INSULÍNICA. 13:05 h
Antonio Muñoz Hoyos, Francisco Rodríguez Argente del Castillo, Gracia M. García Lara, Sergio Muñoz Sánchez, Juan Manuel Fernández García, Carlos Jesús Ruiz Cosano, Antonio Molina Carballo, Irene Machado Casas
Hospital Clínico Universitario San Cecilio, Granada y Universidad de Granada, Granada.
Introducción: Con independencia de la aplicación de protocolos de baja talla en nuestra unidad, estamos especialmente interesados en analizar otros aspectos endocrinometabólicos que acontecen tras la aplicación de determinadas pruebas funcionales. En la presente aportación de datos preliminares pretendemos profundizar en el conocimiento de mecanismos fisiopatológicos que intervienen en tras un test de estímulo hipofisario.
Material: El material objeto del presente estudio está compuesto por una muestra de 15 pacientes con las características de tener una talla por debajo del percentil 3 y estar sometidos al estudio del protocolo de baja talla, que como sabemos necesita en sus últimas etapas realizar al menos dos pruebas de estudio hipofisario. Finalmente parte de estos pacientes fueron diagnosticados de retraso constitucional de crecimiento y baja talla familiar.
Metodología:1) desde el punto de vista metabólico, el aspecto más relevante es la aplicación del protocolo de baja talla de nuestra unidad y la obtención de muestras basal, a los 30', 60', 90', 120', tras la administración de una insulina (0,1 UI/kg), 2) para la determinación de: peroxidación lipídica (espectofotometría).y glutation total, oxidada y reducida (espectofotometría), 3) Finalmente para el análisis estadístico de los datos se utiliza un test de ANOVA de una vía.
Resultados: En los resultados de este estudio se comprueba una disminución en los niveles séricos de los sistemas enzimáticos anti-oxidativos. En el análisis comparativo entre mediciones basales y a los 120 y 90 minutos encontramos con los actuales datos preliminares, glutation total: basal (media 2,35 milimoles/mL ± 1,27) 120' (media 1,68 mmoles/ mL ± 0,69) con una p = 0,033, glutation oxidada: basal(media 1,72 milimoles/mL ± 1,01) 120'(media 1,04 mmoles/ mL ± 0,05) con una p = 0`018, glutation reducido basal (media 0,67 milimoles/mL ± 0,32) 120' (media 0,45 mmoles/ mL ± 0,27)con una p = 0,037: tras el pertinente análisis estadístico, encontramos entre la toma basal y a los 120' diferencias estadísticamente significativas.
Conclusión: Es razonable admitir que el estrés agudo producido por la hipoglucemia insulínica produce claramente un desplazamiento de los sistemas enzimáticos antioxidantes. En consecuencia se hace patente la necesidad de medir la magnitud de este desplazamiento en ulteriores estudios con datos definitivos.
P345 CONVULSIONES NEONATALES COMO PRIMER SIGNO DE HIPERPARATIROIDISMO MATERNO. 13:10 h
Esmeralda Núñez Cuadros, Celia Gómez Robles, Ana M. Cordón Martínez, Mª Ángeles Tejero Hernández, Mateo Arana Aguera, José López López, Juan Pedro López Siguero, Antonio Jurado Ortiz
Hospital Materno Infantil Carlos Haya, Málaga.
Introducción: Las convulsiones en el recién nacido requieren un diagnóstico diferencial amplio que debe incluir aquellas patologías que puedan tener su origen en causas maternas.
Caso clínico: Recién nacida mujer de 13 días de vida que presenta cuadro de 4 días de evolución caracterizado por sacudidas musculares breves, repetitivas, erráticas, de las 4 extremedidades de segundos de duración en número de 4-6/día. Antecedentes Personales: Embarazo controlado, parto vaginal eutócico, RNAT (37 semanas) PAEG (2.950 gr). La exploración era normal destacando la presencia de un cefalohematoma parietal derecho de 3 x 4 cm. Pruebas complementarias: hemograma normal, bioquímica con Calcio total: 6 mg/dl, magnesio: 1,1 mg/dl y fósforo: 10,5 mg/dl, equilibrio acido-base normal, eco de cráneo normal, EEG: bajo voltaje de la actividad bioeléctrica de base, índice calcio/creatinina en orina elevado, PTH: 11 pg/ml y 1,25 (OH)2 vitamina D: 22 pg/ml. Ante estos datos se confirma la presencia de un hipoparatiroidismo. Para descartar la presencia de un Síndrome de Di George que cursa con este trastorno bioquímico se realizaron otras pruebas complementarias como eco de timo que confirmó la presencia de este último, eco abdominal, estudio cardiológico y oftalmológico que fueron normales. Sospechando patología materna se realizó bioquímica: calcio: 11,5 mg/dl, fósforo: 2,0 mg/dl y PTH: 93 pg/ml. De esta forma se confirma el hiperparatiroidismo materno como causa del hipoparatiroidismo neonatal. Mediante gammagrafía con tecnecio se objetiva la presencia de un adenoma paratiroideo en la madre que precisará tratamiento quirúrgico. Durante el ingreso hospitalario recibió tratamiento con gluconato cálcico y sulfato magnésico al 15% iv junto con vitamina D3, pasándose posteriormente a vía oral con normalización de los parámetros bioquímicos 3 meses después.
Comentarios: Las convulsiones neonatales pueden provocar lesiones neurológicas irreversibles, de ahí la necesidad de un diagnóstico precoz para mejorar el pronóstico. La hipocalcemia neonatal puede ser la primera manifestación de una enfermedad materna como el hiperparatiroidismo cuya causa más frecuente es el adenoma paratiroideo. En la literatura existen pocos casos clínicos similares de tan buen pronóstico.
P346 FACTORES PREDICTORES DE TALLA ADULTA EN HIPERPLASIA SUPRARRENAL CONGÉNITA. 13:15 h
Paloma Cabanas Rodríguez, Tania Arévalo Saade, José M. Iglesias Meleiro, Lidia Castro Feijoo, Jesús Barreiro Conde, Lourdes Loidi Fernández de Trocóniz, Celsa Quinteiro García, Manuel Pombo Arias
Hospital Clínico Universitario, Santiago de Compostela (A Coruña).
Introducción: La hiperplasia suprarrenal congénita (HSC) por déficit de 21-hidroxilasa es un trastorno de la estereidogénesis suprarrenal. El aumento de andrógenos puede ocasionar una maduración ósea prematura, asociado a aumento de la velocidad de crecimiento inicial, pero puede llevar a una reducción en la talla adulta. Por otro lado los glucocorticoides exógenos afectan al crecimiento.
Objetivo: Obtener información de la relación existente entre la talla adulta, los posibles factores, como la dosis de glucocorticoides y el grado de supresión hormonal, y así optimizar el tratamiento.
Métodos: Se estudiaron 21 pacientes diagnosticados de HSC, relacionando el crecimiento y talla, con los datos clínicos, hormonales y el genotipo específico a cada paciente.
Resultados: Se distribuyeron en grupos los pacientes estudiados, considerando su SDS de talla final. Ver tabla.

Conclusiones:a) El SDS de talla final hallado en nuestros pacientes no es significativamente inferior al SDS de talla genética. b) Así mismo, los distintos grados de severidad de las mutaciones halladas no se relacionan con la talla final. c) En los pacientes con talla final menor la diferencia entre la edad ósea (Eo) y la edad cronológica (Ec) al diagnóstico es mayor. d) Los niveles de andrógenos deben usarse conjuntamente con la velocidad de crecimiento para optimizar la dosis de glucocorticoides. Así, para alcanzar una mayor talla adulta en pacientes con HSC parece adecuado aconsejar la menor dosis eficaz de hidrocortisona.
HEMATOLOGÍA
P347 LEUCEMIA AGUDA LINFOBLÁSTICA: NUESTRA CASUÍSTICA. 12:00 h
Pedro Cortés Mora, M. Ángeles Vázquez López, Francisco Lendínez Molinos, Moisés Leyva Carmona, M. Ángeles Llamas Guisado, Encarnación López Ruzafa, María Rodríguez Martínez, Purificación Aguilera Sánchez, Raúl Sánchez López, Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
Objetivo: Analizar la evolución seguida por los niños diagnosticados de leucemia aguda linfoblástica en nuestro hospital durante el período comprendido entre Enero-90 y Diciembre-02.
Material y métodos: Se revisan las historias clínicas de 42 pacientes (29 varones y 13 mujeres), edades entre 10 meses y 14 años, diagnosticados de leucemia aguda linfoblástica. Se analizan las características clínicas y analíticas en el momento del diagnóstico, fenotipo inmunológico, alteraciones citogenéticas, respuesta al tratamiento de inducción, índice de riesgo, aparición de recidiva y exitus. Seis pacientes recibieron tratamiento según protocolo BFM y el resto el establecido por la Sociedad Española de Hematología Pediátrica (SHOP). El período de seguimiento medio correspondió a 80,9 ± 49 meses (seguimiento mínimo 12 meses). Se analiza la supervivencia global y los factores predictores y relacionados con la aparición de recidiva y exitus.
Resultados: Motivo de consulta más frecuente: Fiebre (45%); presentaron hepatomegalia el 75,6%, esplenomegalia el 63,4%, afectación del SNC un 4,9% y ósea otro 4,9% (2 casos); presentaron anemia el 88%, trombopenia el 76,2%, > 50.000 leucocitos un 16,7% y > 100.000 un 9,5%. El porcentaje de blastos linfoides en MO correspondió al 90% ± 14%: El 70% fueron catalogados como L2. Los inmunofenotipos más frecuentes fueron el pre-B (46,3%) y común (39%). El 48,8% no presentó alteraciones citogenéticas, un 19,5%(8 casos) si las presentaron (4 hiperploidias, 3 Down, 1 t (4,11). Ocho pacientes (20%) recidivaron entre los 12 y 48 meses del diagnóstico. La supervivencia global a los 96 meses de seguimiento fue del 80%. Ocho pacientes fueron exitus (9 días-45 meses del diagnóstico) Solo la variable hiperleucocitosis resultó significativa (p < 0,01), al analizar las posibles causas de recidiva. Tomando la variable exitus como dependiente, solo la hiperleucoitosis y la presencia de recidiva predijeron el exitus (p < 0,001, y p < 0,000) respectivamente.
Conclusiones: La supervivencia global de nuestra casuística a los 96 meses de seguimiento fue alta. Los únicos factores que se mostraron directamente relacionados con la evolución desfavorable fueron la presencia de hiperleucocitosis y la aparición de recaída.
Otros factores con demostrado poder predictivo (afectación SNC, edad, fenotipo inmunológico, citogenética, respuesta al tratamiento de inducción) no llegaron a tener significación estadística debido probablemente al escaso número de casos.
P348 HISTIOCITOSIS SINUSAL CON LINFADENOPATÍA MASIVA (ENFERMEDAD DE ROSAI DORFMAN): PRESENTACIÓN DE TRES CASOS. 12:05 h
José Manuel Sánchez Granados, Mª Verísima Barajas Sánchez, Olga Serrano Ayestarán, Cristina Ruiz Serrano, Miriam Blanco Rodríguez, Ruth González Crisóstomo, Mercedes Bernacer Borja
Fundación Jiménez Díaz, Madrid.
Introducción: La histiocitosis sinusal con lifadenopatía masiva (HSLM) es una entidad de presentación infrecuente y escasamente documentada en la literatura. Su incidencia es mayor en niños y adolescentes, presentándose usualmente como una masa cervical indolora de curso variable y pronóstico favorable. Presentamos tres casos, uno de ellos asociados a hipogammaglobulinemia
Material y métodos: Se revisaron de forma retrospectiva las historias clínicas de los casos de HSLM diagnosticados en la Fundación Jiménez Díaz, Madrid. El diagnóstico de HSLM se estableció basándose en los hallazgos microscópicos y la positividad a la proteína S-100 con marcador CD1a negativo.

Conclusiones: La HSLM es una enfermedad infrecuente pero a tener en cuenta en el diagnóstico diferencial de las adenopatías, especialmente en las de localización cervical y con afectación extranodal. Debido a su buen pronóstico, en la mayoría de los casos está justificado un tratamiento conservador.
P349 ESFEROCITOSIS HEREDITARIA SIN ANTECEDENTES FAMILIARES. 12:10 h
Sandra Rovira Amigo, Irene Álvarez González, Carolina López Martínez, Patricia Company Maciá, Rosa Garrido Uriarte, Yolanda Ruiz del Prado, M. Luisa Poch Olive, J. Antonio Pérez Marrodan, Alberto Fernández Villamor
Complejo Hospitalario San Millán - San Pedro, Logroño (La Rioja).
Introducción: La esferocitosis hereditaria (EH) es la más frecuente de las anemias hemolíticas hereditarias con una incidencia 1/5000 individuos. Se caracteriza por anemia, ictericia y esplenomegalia con esferocitos en el frotis sanguíneo, reticulocitosis y disminución de la resistencia globular a soluciones salinas hipotónicas.
Caso clínico: Lactante de dos meses con rechazo de las tomas, anemia y aumento del tinte ictérico de 24 horas de evolución. Precisó transfusión de concentrado de hematíes.
Antecedentes personales: RNT. Hiperbilirrubinemia 16,5 mg/dl a las 40 horas de vida que precisó fototerapia, Hb 13,3g%, VCM 95fl. Coombs directo negativo. Grupo sanguíneo de madre e hijo: A positivo.
Antecedentes familiares (AF): No patología sugestiva de crisis hemolíticas y anemia.
Exploración física (EF): Afebril. Tinte ictérico de piel. Abdomen blando, depresible, hepatoesplenomegalia de dos cm. Resto de EF normal.
Pruebas complementarias: Hb 6,1g%, VCM 74fl, HCM 96pg, CHCM 37,1g%, reticulocitos corregidos 2,73%. Abundantes esferocitos y elementos hipocromos en la extensión de sangre periférica. Sistemático de orina normal, no se aprecia hemoglobinuria. Bilirrubina total 5mg% (indirecta 4,9mg%), haptoglobina <6mg%. Sideremia y ferritina normal. Estudio familiar: madre y padre con Hb normal, no esferocitosis, reticulocitos normales.
Evolución: Ante la sospecha de EH se estudia membrana eritrocitaria: G6PDH, PK y electroforesis Hb normales. La resistencia globular osmótica con incubación y el test de autohemólisis salina y con glucosa son indicativos de EH.
Conclusiones: La EH es una entidad con herencia autosómica dominante o recesiva pero podemos encontrar un 10-25% de casos sin AF. En la mayoría de casos, en el período neonatal aparece ictericia con bilirrubina elevada sin paralelismo con la cifra de Hb, que es normal o poco descendida. La disminución de la resistencia globular a soluciones salinas hipotónicas es un test diagnóstico excelente, y la alteración de la prueba de autohemólisis es muy sensible y poco específica.
P350 ALTERACIONES DE LA HEMOSTASIA REFERIDAS A UNA CONSULTA DE HEMATOLOGÍA PEDIÁTRICA. 12:15 h
M. del Mar Guerrero Soler, Paloma Galarón García, M. Elena Cela de Julian, Cristina Beléndez Bieler, M. Ángeles Cantalejo López, Ana Rodríguez-Huertas
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción: Las alteraciones de las pruebas de coagulación son un motivo frecuente de consulta hospitalaria. Aunque en muchas ocasiones son transitorias ó 2ª a un defecto de la técnica, es importante realizar un estudio correcto para descartar una alteración primaria de la hemostasia y, de esta manera, paliar las posibles complicaciones asociadas.
Material y método: Estudio prospectivodescriptivo de pacientes con alteraciones de la hemostasia diagnosticados en la consulta de Hematología Pediátrica del H.G.U Gregorio Marañón durante los años 2002-03. Hemos excluido las 2ª a otras patologías y las púrpuras trombocitopénicas idiopáticas.
Resultados: Se confirmaron un total de 24 casos (18 varones y 6 mujeres) con una edad media al diagnóstico de 10 años (± 2,1 años). El 54% de los pacientes procedían de Atención Primaria y el resto de otras consultas del hospital (12% Anestesia, 16% ORL y el 12% restante de Neuropediatría, Cardiología y Alergia). Sólo 10 niños (42%) tenían clínica (100% sangrado cutáneo-mucoso) y de ellos sólo dos pacientes presentaron complicaciones graves. Respecto a los pacientes asintomáticos (58%), 5 casos fueron detectados de forma casual, 6 casos dentro de estudio familiar y en 3 no se correlacionaba con la clínica presentada. El estudio básico de coagulación mostraba alguna alteración en el 58% de los casos. Entre las alteraciones con tendencia a hemorragia hubo 13 casos de déficit leve de uno o varios factores en rango hemostático (58%), 1 caso de déficit de factor VII moderado y 3 de Enfermedad de Von-Willebrand tipo I. En cuanto a las alteraciones trombóticas 2 casos de déficit leve de proteína S libre, 1 de resistencia familiar a la proteína C, 1 de mutación heterocigota FII-20210 y 1 del factor V de Leyden. Dentro de las trombopatías se diagnosticó un caso de Enfermedad de BernardSoulier y una Trombopenia con alteración funcional autosómica dominante. Detectamos afectación familiar en el 41% de los casos
Conclusiones: 1) La mayoría de las alteraciones encontradas han sido leves y no precisarán tratamiento o sólo lo requerirán ante situaciones de riesgo conocidas. 2) Importancia de realizar un estudio familiar en determinadas alteraciones.
P351 MANIFESTACIONES PSEUDOREUMÁTICAS COMO FORMA DE PRESENTACIÓN DE LEUCEMIA LINFOIDE AGUDA. 12:20 h
Laura San Feliciano Martín, Montserrat Berrocal Castañeda, María Bengoa Caamaño, Susana González de la Gándara, Manuela Muriel Ramos, Dorotea Raquel Fernández Álvarez, Gabriel Mateos Pérez
Hospital Universitario, Salamanca.
La LLA (leucemia linfoide aguda) es hoy la neoplasia más frecuente en la infancia. Hoy en día representa el 30% de todos los cánceres en edad pediátrica y su incidencia alcanza 250 nuevos casos anuales en España. Tiene una alta probabilidad de curación, pero esto depende en gran parte de un diagnóstico precoz. Los síntomas más frecuentes al diagnóstico son: compromiso del estado general, palidez o anemia, adenopatías y fiebre. El papel del pediatra en el diagnóstico es fundamental y más aún en aquellas formas de presentación que simulan enfermedades de otra etiología como los dolores óseos o alteración de la marcha. Presentamos una revisión de 6 casos de LLA en niños cuyas formas de presentación fueron manifestaciones pseudoreumáticas como dolores óseos generalizados o localizados, alteración de la marcha, dolor de tobillo... que en muchos casos llevaron a un retraso en el diagnóstico. Analizamos: edad, sexo, diagnóstico inicial, diagnóstico definitivo, exámenes complementarios realizados y tiempo de demora al diagnóstico.
Comentarios: La observación clínica de nuestros pacientes nos hace destacar la alta frecuencia de presentación de la LLA en forma de dolores óseos erráticos. Resulta de gran importancia la práctica de un estudio hematológico básico de forma precoz en cualquier niño que presente dolores óseos injustificados, así como un seguimiento cercano para retrasar lo menos posible el diagnóstico y el tratamiento quimioterápico.
P352 BICITOPENIA AUTOINMUNE: A PROPÓSITO DE UN CASO. 12:25 h
Ana M. Galera Miñarro, Mª José Romero Egea, Juana M. Espín López, Juan José Quesada López, M. Ester Llinares Riestra, Mar Bermúdez Cortés, José Luis Fuster Soler
Hospital Universitario Virgen de la Arrixaca, Murcia.
Antecedentes y objetivo: La combinación de trombocitopenia y neutropenia autoinmune en muchos casos se describe en asociación a otros procesos como trastornos autoinmunes, infecciones, fármacos, enfermedades malignas o estados de inmunodeficiencia. A diferencia de la púrpura trombocitopénica idiopática (PTI), en estos casos con frecuencia se describe la presencia de hallazgos como linfadenopatía, esplenomegalia y anomalías inmunológicas. Se describe además una alta tasa de fracaso de los tratamientos convencionales para la PTI (corticoides, inmunoglobulinas y esplenectomía). La indicación de la esplenectomía resulta por tanto controvertida en estos casos. Mostramos los hallazgos clínicos y analíticos y la evolución de un paciente diagnosticado de trombocitopenia y neutropenia autoinmune primaria.
Métodos: Presentamos un caso de PTI crónica asociada a neutropenia autoinmune de origen primario. Los recuentos de plaquetas y granulocitos previos al inicio del tratamiento oscilaron entre 92 y 7 x 109/L y 2.2 y 0,8 x 109/L, respectivamente. El examen de anticuerpos antiplaquetarios resultó reiteradamente negativo. Se detectó la presencia de anticuerpos tipo IgG antigranulocito a título fuerte. Otros estudios inmunológicos (incluyendo nivel de inmunoglobulinas, subpoblaciones linfocitarias en sangre periférica, anticuerpos no órgano específicos, test de Coombs directo y anticuerpos antifosfolípido) resultaron negativos. El aspirado medular mostró hiperplasia megacariocítica en ausencia de otras anomalías. Se orientó como PTI crónica asociada a neutropenia autoinmune.
Resultados: El proceso evolucionó como una PTI crónica moderada durante 12 meses sin que se administrara ningún tratamiento. Posteriormente mostró recuentos plaquetarios inferiores a 20 x 109/L, comportándose como corticodependiente por lo que se indicó tratamiento secuencial con gammaglobulinas intravenosas. Tras 12 meses de tratamiento se indicó la esplenectomía, comprobándose la resolución de la trombopenia y permaneciendo estable el recuento de granulocitos.
Conclusiones: La combinación de PTI asociada a neutropenia autoinmune puede presentarse de forma aislada. En nuestro caso, la ausencia de hallazgos clínicos y analíticos asociados podría interpretarse como factor predictivo de buena respuesta a la esplenectomía.
P353 DETECCIÓN PRECOZ DE HEMOCROMATOSIS EN PEDIATRÍA: CUÁNDO SOSPECHARLA. 12:30 h
Sonia Villar Castro, M. del Mar Guerrero Soler, M. Elena Cela de Julian, Cristina Beléndez Bieler, Paloma Galarón García, M. Ángeles Cantalejo López, Raquel Navajas
Hospital General Universitario Gregorio Marañón, Madrid y Laboratorio de Estudios Genéticos Megalab, Madrid.
La hemocromatosis hereditaria (HCH) es una enfermedad autosómica recesiva frecuente en la raza caucásica, a menudo infradiagnosticada por la ausencia e inespecificidad de los síntomas iniciales. Las mutaciones conocidas en el gen HFE son C282Y, H63D y S65D, destacando por su incidencia el genotipo C282Y en homocigosis (90%) y la mutación C282Y/H63Y (5%), seguida de la H63D en heterocigosis (4%) y homocigosis (2%) y C282Y en heterocigosis (1-2%).
Objetivo: Comunicar los casos de hemocromatosis vistos en la consulta de Hematología en 1 año para conocer mejor su diagnóstico en la edad pediátrica.
Métodos: Revisión de los casos diagnosticados de hemocromatosis en el año 2003.
Caso 1: niña de 10 años asintomática, enviada por su pediatra al detectar en analítica rutinaria una ferritina de 700 ng/ml con el resto de los parámetros normales. EF normal. AF: abuelo paterno diabetes, abuela materna cirrosis hepática Ante este hallazgo se realizó diagnóstico diferencial de hiperferritinemia (infecciones, anemia megaloblástica o hemolítica, enfermedad hepática crónica, enfermedades autoinmunes) descartándose por anamnesis transfusiones recientes, dieta rica en hierro o ingesta de fármacos. Se solicitó estudio genético de HCH detectándose la mutación H63D en heterocigosis.
Caso 2: hermanas de 5 y 8 años enviadas por padre diagnosticado de hemocromatosis (mutación H63D en homocigosis). Asintomáticas. EF normal. AF: abuelo paterno IAM, abuela materna pancreatitis crónica. Hemograma y ferritina normales. Estudio genético de HCH: mutación H63D en heterocigosis.
Caso 3: niña de 8 años, enviada por su pediatra por hermana diagnosticada de hemocromatosis (mutación H63D en heterocigosis) tras encontrarse ferritina elevada en análisis rutinario. Asintomática. EF normal. Hemograma y feritina normales. Estudio genético de HCH: madre C282Y en heterocigosis, padre H63D en heterocigosis. Hermana H63D en heterocigosis. Paciente C282Y en heterocigosis
Conclusiones:1) La HCH es la forma más frecuente de enfermedad producida por el depósito de hierro, debiéndose realizar estudio genético cuando existe historia familiar de hemocromatosis o ante el hallazgo de niveles elevados de ferritina. 2) La mayoría de los pacientes con HCH son homocigotos para la mutación C282Y pero solo un pequeño porcentaje desarrollarán clínicamente la enfermedad.
P354 RECIÉN NACIDO CON DÉFICIT DE ANTITROMBINA III. 12:35 h
Ana Mª Pérez Aragón, María José Gutierrez Pimentel, Manuel Samaniego Muñoz, Ana Rosa López-Quiñones Pimentel, Catalina González Hervás, M. Fernanda Moreno Galdó, Mauricio Conde Otero, Francisco García Róspide, Manuel Jurado Chacón, Luis Moltó Ripoll
Hospital Virgen de las Nieves, Granada.
Introducción: Los síndromes trombofílicos congénitos son raros, constituyendo un problema en cuanto a morbi-mortalidad. No disponemos de casuísticas que permitan estudios sobre prevención primaria, secundaria, y líneas de tratamiento sustitutivo. En el sistema hemostático, son elementos de predicción de riesgo de trombosis las deficiencias hereditarias de AT III y proteína C.
Caso clínico: Recién nacido varón, de 15 meses de edad que al nacimiento presentaba una lesión roja en cara anterior de antebrazo izquierdo, 3 x 1 cm.
AF y P: Déficit de AT III y factor V de Leyden en la familia materna, con consanguinidad entre abuelos. Madre con ambos déficits, diagnosticados tras la gestación, siendo homocigota para la mutación del gen de la metilentetrahidrofolato reductasa en la posición 677, sin antecedentes trombóticos. Embarazo controlado y parto inducido por sufrimiento fetal agudo. LA teñido y vuelta de cordón con nudo verdadero. APGAR 3-7-9, reanimación III..A las 14 horas de vida la lesión ocupaba toda la cara anterior de antebrazo y mano e impresionaba de patología vascular sugerente de isquemia arterial aguda de origen trombótico. En la exploración clínica se apreciaba ausencia de pulsatilidad axilar, humeral y distal, confirmando el estudio con Eco-Doppler la trombosis a nivel axilo-subclavio-humeral izquierda de todo el eje. Los hallazgos con Eco-Doppler explicaron que, al no tener colateralidad de suplencia suficiente fuera ampliándose la zona de prenecrosis desde los dedos hasta la mitad del antebrazo en las 24-48 horas siguientes. Estudio de coagulación: actividad de protrombina: 25%, INR: 3,2; APTT: 56,6; AT III: 36%. Confirmado el déficit, se inició tratamiento con Kybernin-P y heparina sódica a dosis anticoagulantes, lográndose cifras de APTT:30% y AT III: 70%, mejorando la perfusión de brazo y tercio superior del antebrazo con necrosis y amputación de la mitad inferior de antebrazo y mano.
Conclusiones:1) El embarazo y parto en una deficiencia congénita de ATIII, presenta alto riesgo de trombosis y requiere medidas profilácticas con ATIII, 2) La trombosis arterial es una afección rara que hay que tener en cuenta ante cualquier síndrome trombofílico congénito, 3) Creemos que la ATIII debe ser medida en los recién nacidos con historia familiar de déficit y en aquellas situaciones con procesos trombóticos inexplicables.
P355 TROMBOSIS DEL SENO VENOSO EN UN NEONATO: A PROPÓSITO DE UN CASO. 12:40 h
Pablo Sáez Pérez, Cristina Serra Amaya, Pere R. Balliu Badía, Mario J. Sánchez Fernández, Alberto Trujillo Fagundo, Emma Amatller Malfaz, Carme González Mancilla, Gemma Giralt García, Josep M. Mengibar Garrido, Jaume Macià Martí
Hospital Universitario Dr. Josep Trueta, Girona.
Objetivo: Exposición de un nuevo caso de trombosis del seno venoso en un neonato de 25 días de vida.
Caso clínico: Neonato de 25 días de vida, sin antecedentes patológicos de interés, que presentaba hipoactividad acompañada de vómitos alimentarios sin productos patológicos y alguna deposición dispéptica sin productos patológicos, manteniéndose afebril. Peso 3.200 gr (pérdida de 100 gr respecto al nacimiento). Mal estado general. Distrófico. Hipoactivo. Mucosas secas. Signo del pliegue positivo. Peristaltismo aumentado. Hb 12 g/dl, Hto 34%, VCM 97,6 fL, leucocitos 19.800 K/mcL (1% promielocitos, 2% metamielocitos, 8% cayados, 28% neutrófilos, 47% linfocitos, 12% monocitos). Sodio 130,6 mEq/L. PCR 1.4 mg/dL. Tasa protrombina 66%, Fibrinogeno 106. Se inició sueroterapia endovenosa. Pocos minutos después de su ingreso en la UCI-Pediátrica presentó convulsión tónica. Se realizó punción lumbar hallándose LCR hemático, cuyo cultivo fue negativo. Se efectuó ecografía cerebral y RMN craneal que mostraron trombosis aguda de sistema venoso superficial (seno longitudinal superior) y profundo, con presencia de un infarto talámico izquierdo y edema talámico bilateral y hemorragia tetraventricular con severa hidrocefalia. Ante dichos hallázgos se inició heparina sódica, pasándose posteriormente a heparina de bajo peso molecular. Se colocó drenaje ventricular externo que se mantuvo durante diecisiete días. Se efectuó estudio de coagulación en que se detectó Antitrombina III 42%, Proteína C 23%, Proteína S 57%. Se apreció estancamiento ponderal y persistencia de deposiciones dispépticas mostrando en el RAST a proteínas de leche de vaca Ig E en el nivel alto por lo que se inició hidrolizado de proteínas cediendo la sintomatología. Previamente al alta hospitalaria se efectuó nueva RMN craneal que mostró persistencia de hemorragia tetraventricular con corrección parcial de la severa hidrocefalia, recanalización parcial del seno longitudinal superior a nivel de su tercio inferior con ausencia de flujo en su tercio medio y anterior, y recuperación del flujo en el sistema venoso profundo. Actualmente está pendiente de estudio familiar y nueva determinación de proteína C.
Discusión: La trombosis del seno venoso infrecuente en la infancia, afectando principalmente a neonatos. Un porcentaje importante presentan coagulopatías. En nuestro caso la causa probables fue un déficit de proteína C.
P356 HEMATOMA INTRAESPINAL EN UN NIÑO CON HEMOFILIA GRAVE Y TÍTULO ALTO DE INHIBIDORES. 12:45 h
Miguel Lillo Lillo, Montserrat Maicas Mascarell, Ester Gil Pons, Ana Pérez Pardo, Carolina Gutiérrez Junquera, Rafael Ruiz Cano, Alberto Vidal Company
Complejo Hospitalario - Hospital General, Albacete.
Introducción: El sangrado intraespinal es una complicación rara de la Hemofilia y representa entre el 2 y el 8% de las hemorragias en el sistema nervioso central de estos enfermos. Las manifestaciones clínicas incluyen dolor regional o radicular, debilidad muscular, parálisis y alteraciones sensitivas, que varían según la gravedad de la compresión medular. Presentamos un niño diagnosticado de Hemofília A grave, y títulos elevados de inhibidores con tetraparesia secundaria a hematoma epidural extenso.
Caso clínico: Varón de 3 años de edad que ingresa por presentar irritabilidad e impotencia funcional de EEII. Estaba diagnosticado de Hemofilia A grave desde el primer año de vida. Había precisado varios ingresos por diversas complicaciones hemorrágicas: en escroto y cérvico-facial, entre otras. Había recibido tratamiento con Factor VIII recombinante y había desarrollado inhibidores a título elevado. En la anamnesis se deduce el antecedente de un traumatismo sobre el tórax. Llama la atención en la exploración física la presencia de paresia de EEII y posteriormente afectación motora de EESS. No existía compromiso cardiorrespiratorio ni hemodinámico. En la RMN se apreció lesión hipodensa que abarcaba desde C4 hasta sacro correspondiente a hematoma epidural. En C4-C5 y en D5-D6 había lesiones intramedulares de características isquémicas. Recibió tratamiento con corticoides, F VIIa y control de Rehabilitación en unidad especializada. La evolución posterior ha sido buena, con recuperación paulatina de sus funciones neuromotoras, si bien persiste discreta paresia en EEII.
Comentarios: La presencia de procesos hemorrágicos intraespinales confiere una gravedad importante a las complicaciones esperadas en un niño pequeño con Hemofilia grave. Algunos de los pacientes publicados en tales circunstancias llegan a un desenlace fatal por la facilidad de repercusiones cardiorrespiratorias e indirectamente infecciosas. El empleo precoz del tratamiento oportuno permite augurar un mejor pronóstico, como ocurrió en el niño que presentamos.
P357 PÚRPURA CONGÉNITA DE ETIOLOGÍA INFRECUENTE. 12:50 h
Begoña Martínez Pineda, Raquel Perera Soler, Ricardo López Almaraz, José Cayetano Rodríguez Luis, Cristina León Quintana, Roque Abián Montesdeoca Melián, Judith Mesa Fumero
Hospital Universitario Materno-Infantil de Canarias, Las Palmas.
El síndrome purpúrico congénito por defecto de la función plaquetaria es una patología rara en nuestro medio. Presentamos un caso de púrpura presente desde el nacimiento de etiología infrecuente.
Nota clínica: Varón de 19 meses de edad, que acude a consulta de Hematología pediátrica, por presentar desde el nacimiento hematomas generalizados ante mínimos traumatismos, epistaxis y gingivorragia ocasional, sin relación con ingesta medicamentosa, ni enfermedad asociada.
Como antecedentes personales destaca la presencia de un síndrome purpúrico generalizado a las pocas horas de vida con hematomas múltiples en tórax, abdomen, región cervical y periauricular, petequias diseminadas por toda la economía y hemorragia digestiva alta, sin sangrados a otros niveles. Aportaba hemograma y coagulación básica (TP, APTT, fibrinógeno) repetidamente normales.
Como antecedentes familiares existe consanguinidad entre los padres, y un familiar de 2º grado con episodios de sangrado, ante traumatismos leves, no filiado hasta la fecha.
Se realizan los siguientes exámenes complementarios: a) Hemograma con recuento y morfología plaquetaria: 310.000/mm3, bioquímica con función renal y hepática dentro de la normalidad, b) Tiempo de protrombina: 100%, APTT: 31 seg, fibrinógeno, dímero-D, antitrombina III y factor von Willebrand: normales.Tiempo de hemorragia > 300 seg.
Ante la sospecha de trombopatía se realiza estudio de agregación plaquetaria que resulta ausente al ADP, adrenalina y colágeno, pero presente con ristocetina.
Conclusiones: Ante un síndrome purpúrico congénito, en el contexto de enfermedad familiar, con un tiempo de hemorragia alargado y sin disminución del número de plaquetas debemos descartar un defecto del funcionamiento plaquetario (Trombopatía). Si el estudio de la función plaquetar "in vitro" presenta alteración de la agregación es sugestivo de Tromboastenia de Glazmann, que se confirma con la medición de los niveles de la glicoproteína IIb-IIIa y el estudio genético.
P358 SÍNDROME HEMOFAGOCÍTICO SECUNDARIO A INFECCIÓN POR VIRUS DE EPSTEIN BARR. 12:55 h
José Camino López Pena, Isabel Badell Serra, Bibiana Pineda Prats, Almudena Sánchez Vázquez, Gemma Arca Díaz, Antonio Torres Culell, Eduard Carreras González, M. Adela Retana Castán, Juan Nadal Amat, José Cubells Rieró
Hospital de la Santa Creu y Sant Pau, Barcelona y Universidad Autónoma, Barcelona.
Introducción: El síndrome hemafagocítico (SHF) es una entidad nosológica poco frecuente y heterogénea, con alta morbimortalidad. Forma parte de las histicitosis de clase II distinguiéndose una forma familiar, con herencia autosómica recesiva, y otra secundaria a enfermedades subyacentes especialmente infecciones víricas.
Caso: Varón de 12 meses, sin antecedentes de interés, que consulta por cuadro febril y dificultad respiratoria de 5 días de evolución, en tratamiento con Amoxicilina-ácido clavulánico. EF: mal estado general, destaca dificultad respiratoria franca, hepatomegalia de 1cm, polo de bazo y poliadenopatías. Analítica:Hb: 84g/l, VCM: 79f/l, Leucocitos:26.300/mm3 (60N/25L/5M/6Bd/4Meta/linfomonocitarias), plaquetas: 365.000/mm3. LDH: 1067 U/L. Rx tórax: infiltrado parahiliar derecho. Se inicia tratamiento broncodilatador y Cefotaxima ev. De forma progresiva presenta empeoramiento respiratorio que requiere conexión a ventilación mecánica. Hepatomegalia de 4-5 cm y esplenomegalia de 3cm. Ecografía abdominal hepatoesplenomegalia homogénea sin lesiones focales, resto normal. En controles analíticos destacan anemia normocítica, plaquetopenia, hipoalbuminemia, hipofibrinogenemia, hipertrigliceridemia y hipocolesterolemia. Serologías víricas negativas incluida Virus de Epstein Barr (VEB). Ante la sospecha de SHF se realiza aspirado medular: médula reactiva con abundantes células plasmáticas, resto normal. Punción lumbar: glucosa 6,1 mmol/l, proteínas: 54 g/l, células: 2133 cel/ul con 70% linfocitos y 30% macrófagos con fenómenos de hemofagocitosis. Cultivos negativos. Biopsia hepática: parénquima de estructura normal con infiltrado portal con abundantes células plasmáticas y linfocitos reactivos. Se aíslan linfocitos con proteína latente de membrana tipo I para VEB. PCR para VEB positivo en hepatocitos. Se inicia tratamiento con Dexametasona, VP-16, Ciclosporina A e intratecal, según protocolo. Fallo multiorgánico progresivo y es éxitus a los 40 días.
Conclusión: Ante un cuadro febril, hepatoesplenomegalia, hipofibrinogenemia y citopenia debemos tener en cuenta el SHF, ya que su diagnóstico y tratamiento precoz pueden cambiar su infausto pronóstico. Su forma primaria cuando se asocia a infección por VEB hace necesario plantear el diagnóstico diferencial con el Síndrome Linfoproliferativo ligado al cromosoma X o Enfermedad de Duncan.
P359 FAVISMO EN UN NIÑO DE 13 AÑOS CON ANEMIA HEMOLÍTICA GRAVE. 13:00 h
Yolanda Lage Alfranca, Alejandro López Escobar, Miriam Centeno Jiménez, José María de Cea Crespo, Isabel Pinto Fuentes, M. Luz Cilleruelo Pascual
Hospital Severo Ochoa, Leganés (Madrid).
El déficit de G6P-DH es la enzimopatía eritrocitaria más frecuente y predomina en la raza negra, asiática y en la cuenca mediterránea. Este déficit provoca un descenso de la capacidad antioxidante y un aumento de la sensibilidad eritrocitaria al efecto de ciertos fármacos y alimentos. Se presenta el caso clínico de un varón de 13 años que acude a urgencias por cuadro de mareo con caída al suelo y pérdida de conciencia de unos segundos de duración sin otros datos de focalidad neurológica. Refiere ingesta el día previo de habas (era la segunda vez en su vida que las tomaba). Entre los antecedentes personales destaca en la madre un transplante renal secundario a púrpura de Schonlein-Henoch y cuadros de anemia no filiada durante la infancia. A la exploración física presenta regular estado general con palidez cutáneomucosa e ictericia conjuntival y dolor a la palpación en hipocondrio derecho. El resto de la exploración era normal. En la analítica de urgencias se objetiva anemia normocítica y normocrómica con anisopoiquilocitosis y reticulocitosis asi como hiperbilirrubinemia y aumento de los niveles de haptoglobina sérica. Ante el cuadro hemolítico y la ingesta previa de habas se realiza estudio enzimático detectándose un déficit de G6P Deshidrogenasa. Durante el ingreso presenta empeoramiento clínico con aumento de la palidez cutánea y del dolor abdominal, comprobándose una anemización progresiva (cifras mínimas de Hb 5.5 y HTO 16%) que precisó transfusión de dos concentrados de hematíes con mejoría posterior. Durante el ingreso se realiza estudio enzimático a los padres siendo normal el del padre y detectándose un déficit parcial de G6P-DH en la madre. Como comentario final se debe considerar el diagnóstico de déficit de G6P-DH en cualquier niño que experimenta un proceso hemolítico agudo sobre todo en los procedentes de África ó del área mediterránea, interrogando acerca de posibles agentes oxidantes. Debido a la inmigración en España, nos enfrentaremos más a menudo a casos de este tipo si bien no suelen ser tan graves como éste.
P360 RECTORRAGIA COMO FORMA DE PRESENTACIÓN DE SÍNDROME DE WISKOTT-ALDRICH. 13:05 h
Margarita Cañellas Fuster, Queralt Soler Campins, Nieves Nieto del Rincón, Mercedes Guibelalde del Castillo, Nuria Matamoros, Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca (Baleares).
Introducción: El Síndrome de Wiskott-Aldrich (WAS) es una inmunodeficiencia primaria recesiva ligada X, caracterizada por infecciones repetición, hemorragias y eccema. El diagnóstico se apoya en la clínica, trombopenia periférica (volumen plaquetar bajo), inmunología (disminución IgM e IgG, aumento IgA, IgE y CD4/CD8), y se confirma por Western Blot (ausencia proteína WAS) y estudio genético (gen Xp11.22-11.3). Además del tratamiento de sostén, la reconstitución Stem Cell es el único curativo. La historia natural es de muerte entre la 1ª y 2ª década.
Caso clínico: RN fruto de gestación sin incidencias (serología infecciosa negativa), parto eutócico (PAEG). Lactancia materna. Madre 36 años, sana (G/A/V:3/1/2). Padre 45 años, sano. Hermano 6 años, sano. Consultan al 3er mes, por rectorragias de 2 meses de evolución. Exploración física: Peso 6300g (P 75), Talla 63 cm (P 90). Dermatitis en cara, resto normal. Exploraciones Complementarias: Leucocitos normales con eosinofilia, anemia microcítica, plaquetopenia (66.000 con volumen plaquetario de 7); Serología CMV, VEB, Toxoplasma, VIH negativos; CMV orina negativo; coprocultivos-parásitos negativos. Radiología: Eco abdominal, cerebral y serie esquelética normales. Rectoscopia: mucosa friable, lesiones petequiales e infiltración eosinofilica. Inmunología: linfopenia T CD8; inmunoglobulinas normales; Rast y Prick PLV negativo. Médula ósea: megacariocitos normales. Se confirma la sospecha por Western Blot (disminución proteína WAS) y estudio molecular (delección A 266/267). Madre portadora de la misma mutación. Recibe corticoides (dosis bajas), inmunoglobulinas y profilaxis antibiótica. Presenta rectorragias ocasionales y no ha tenido infecciones relevantes. Se mantiene en búsqueda de donante no emparentado para futuro transplante de progenitores hematopoyéticos (TPH).
Comentarios:1) El WAS es una entidad rara, que debe ser sospechada ante la triada infección, hemorragia y eccema. 2) En nuestro paciente la rectorragia fue el síntoma guía para el diagnóstico. 3) El diagnóstico precoz y su confirmación genética, son importantes para mejorar el pronóstico y planificar el TPH. 4) Aunque el único tratamiento curativo es TPH, un adecuado manejo del paciente es fundamental para evitar complicaciones futuras.
P361 ANEMIA HEMOLÍTICA AUTOINMUNE SECUNDARIA A INFECCIÓN RESPIRATORIA. 13:10 h
Manuel Vázquez Donsion, Sonia Marcos Alonso, José Miguel Couselo Sánchez, Antonio Rodríguez Núñez, José Angel Porto Arceo, Gema Ariceta Iraola
Hospital Clínico Universitario, Santiago de Compostela (A Coruña) y Universidad de Santiago de Compostela, A Coruña.
Introducción: La anemia hemolítica autoinmune en la edad pediátrica es poco frecuente. Comunicamos 2 casos de crisis hemolítica coincidiendo con infección respiratoria.
Casuística:Caso nº 1: Varón de 4 años. Caso nº 2: Varón de 6 años

Evolución: Se hizo tratamiento con prednisona en los 2 casos. Ninguno recibió transfusiones de concentrado de hematíes. El valor mínimo de hemoglobina en el caso 1 fue de 7,4 gr/dL con normalización de la misma a los 10 días; en el caso 2, la hemoglobina disminuyó hasta 7,1 gr/dL y se normalizó a los 30 días. El caso 1 presenta desde el ingreso manifestaciones de glomerulonefritis crónica mesangiocapilar sin insuficiencia renal y el caso 2 una insuficiencia renal aguda leve secundaria a crisis hemolítica con resolución completa.
Comentario: Es destacable, en el primer caso, la asociación de dos enfermedades autoinmunes (glomerulonefritis mesangiocapilar y anemia hemolítica autoinmune) y, en el segundo caso, la coincidencia ya conocida de infección por micoplasma y crisis de hemólisis intravascular.
P362 TROMBOCITOPENIA NEONATAL ALOINMUNE: PRESENTACIÓN DE UN CASO. 13:15 h
Ana Navarro Dourdil, Israel Ordóñez Medina, J.M. Vagace Valero, R Parra, R. Hernández, Juan José Cardesa García
Hospital Materno Infantil, Badajoz.
Introducción: La trombocitopenia fetal/neonatal aloinmune (TNA) es la causa más común de tromocitopenia grave en el recién nacido (1 de cada 1.000 recién nacidos). Se produce por la acción de un aloanticuerpo plaquetario específico materno que reacciona con un antígeno de las plaquetas fetales/neonatales heredado del padre que conduce a la destrucción de éstas. Es un proceso potencialmente muy grave con desarrollo de hemorragia cerebral en un 10-30% de los recién nacidos, secuelas neurológicas irreversibles en un 20% de los casos y mortalidad del 10%.
Caso clínico: Recién nacido hembra de dos horas de vida que presenta exantema petequial generalizado. Padres jóvenes y sanos. Embarazo controlado de 37semanas de curso normal, incluyendo recuento plaquetario. Primigesta. Parto eutocico. Apgar 9/10. Exploración: buen estado general, destaca la abundancia de elementos petequiales generalizados. No presenta aspecto séptico. Resto normal. Exámenes complementarios: plaquetopenia severa (13.000 plaquetas/mm3), serie roja y blanca normales. Estudio de coagulación: normal. Eco cerebral: hemorragia intraventricular sin dilatación de ventrículos. Resto de pruebas complementarias normales. Con la sospecha clínica de TNA, se inicia tratamiento con Inmunoglobulina IV (1 gr/kg/dia, durante 2 días) y transfusión de 1u de plaquetas con respuesta lentamente progresiva, (120.000 plaquetas/mm3 al alta). Control Eco cerebral: normal. Se confirma el diagnóstico tras detectar en suero materno aloanticuerpo de especificidad anti HPA-1a.La determinación del genotipo plaquetario por PCR demostró la imcompatibilidad antigénica maternofetal: RN HPA1a/1b, madre HPA1b/1b, padre HPA1a/1b. Evolución favorable.
Conclusiones: Ante recién nacido con plaquetopenia severa, buen estado general y plaquetas maternas normales, se sospechará TNA iniciándose tratamiento sin dilación. El aloanticuerpo anti HPA-1a es el responsable en el 75-85% de los casos, siendo el de mayor riesgo de hemorragia cerebral. El 30% ocurre en la primera gestación (50% intraútero). Recidiva: 85-90%. La magnitud del problema justificaría la implantación de un programa profiláctico antenatal, pero no existen estudios aleatorizados sobre la relación coste-beneficio del cribado sistemático de gestantes HPA-1a negativo. El tratamiento antenatal se basa en transfusiones intraútero de plaquetas HPA compatibles o la administración de IgGev y/o corticoides a la madre.
P363 COMPLICACIONES INFRECUENTES DEL TRANSPLANTE DE PROGENITORES HEMATOPOYÉTICOS. PRESENTACIÓN DE UN CASO. 13:20 h
María Garatea Rodríguez, Beatriz Solís Gómez, Fidel Gallinas Victoriano, Marian Ardanaz, Joaquín Duarte Calvete, M.A. Díaz, Francisco Javier Molina Garicano
H. Virgen del Camino, Pamplona (Navarra) y H. del Niño Jesús, Madrid.
Introducción: La aplasia medular es una pancitopenia en sangre periférica debida a disminución o ausencia de progenitores hematopoyéticos en médula ósea. El tratamiento de elección de la aplasia grave es el transplante alogénico de progenitores hematopoyéticos con terapia inmunosupresora administrada tanto antes como después del mismo.
Caso clínico: Niño de 14 años diagnosticado de aplasia medular grave en Noviembre de 2002. En Mayo de 2003 se realiza acondicionamiento y transplante alogénico de progenitores hematopoyéticos de donante familiar idéntico, tras lo cual comienza tratamiento con ciclosporina, metil prednisolona y metotrexate, y posteriormente al injerto plaquetar, con cotrimoxazol. En julio de 2003 comienza tratamiento con ganciclovir durante 21 días tras encontrar antígeno de CMV en cultivo virológico de sangre. En agosto de 2003 refiere disminución de agudeza visual de ojo derecho, apreciándose lesión blanquecina que afecta a mácula en la exploración oftalmológica. Ante estos hallazgos se plantea el diagnóstico diferencial. Más tarde se confirmó la serología positiva a toxocara canis.
Discusión: El TPH alogénico es un procedimiento que cada vez es más frecuentemente empleado para tratamiento de algunas patologías pediátricas y que condiciona una importante inmunosupresión y todas las complicaciones que se derivan de ésta. Ante una disminución de la agudeza visual en un niño inmunodeprimido hay que tener en cuenta infecciones parasitarias y entre ellas el toxocara canis.
P364 SÍNDROME HEMOLÍTICO URÉMICO ASOCIADO A NEUMONÍA NECROTIZANTE. A PROPÓSITO DE UN CASO. 13:25 h
Paz Chimenti Camacho, Kay Boris Brandstrup Azuero, Carmen Fernández García-Abril, Amaya Bustinza Arriortua, Augusto Luque de Pablos, Jesús Lopez-Herce Cid
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción: El síndrome hemolítico urémico (SHU) es la causa más frecuente de insuficiencia renal aguda parenquimatosa en el niño. Se caracteriza por anemia hemolítica microangiopática, trombocitopenia e insuficiencia renal aguda. El 90% aparece tras una diarrea infecciosa aguda, generalmente por Escherichia coli 0157:H7 productora de verotoxina; el otro 10% se asocia a múltiples etiologías, sin diarrea y mal pronostico (SHU atípico).
Caso clínico: Niño de 3 años trasladado con el diagnóstico de neumonía bilateral con derrame pleural izquierdo, en tratamiento con amoxicilina-clavulánico. Se cambia antibioterapia empírica a cefotaxima y clindamicina i.v. Se coloca tubo de drenaje torácico obteniéndose escaso líquido hematico. En TAC helicoidal se objetiva neumonía necrotizante, paquipleuritis y derrame pleural izquierdo organizado, precisando decorticación pulmonar y colocación de dos drenajes pleurales. A las 12 horas del ingreso presenta empeoramiento clínico con aumento de la dificultad respiratoria, anemización secundaria a hemólisis (Hb: 4,3 gr/dl; bilirrubina 3,6 g/dl, indirecta 2,8 g/dl, esquistocitos), plaquetas 13.000, leucocitos 17.200 (granulocitos 88%), oligoanuria con hematuria y proteinuria, creatinina 1,4 g/dl (máxima 3,9 g/dl), urea 129 mg/dl (máximo 180 mg/dl) y edemas. Precisó ventilación mecánica 13 días (6 días con ventilación de alta frecuencia), con oxido nítrico inhalado, diálisis peritoneal durante 15 días, soporte inotrópico con dopamina (10 mcg/kg/minuto), tratamiento antihipertensivo con nifedipino y labetalol y transfusión de PFC, plaquetas y concentrado de hematíes lavados por sospecha de SHU asociado a enfermedad neumocócica invasiva. A los 4 días se sustituye tratamiento antibiótico por linezolid que se mantiene 14 días con buena evolución. Todos los cultivos (hemocultivo, líquido pleural, secreciones) fueron estériles. A los 3 meses del episodio presenta una función renal normal.
Comentarios: El SHU atípico puede tener una evolución más grave que el típico, con mayor necesidad de técnicas de depuración extrarrenal y mayor probabilidad de insuficiencia renal a largo plazo. Si se sospecha SHU secundario a enfermedad neumocócica invasora es necesario la administración de derivados sanguíneos lavados.
P365 HEMOFILIA A: DIAGNÓSTICO CASUAL EN UN LACTANTE DE 4 MESES. 13:30 h
Zulema Hernando Zarate, Leire García Sarriugarte, Aitor Ruano López, Ana Vereas Martínez, Agustín Rodríguez Ortiz, José M. Indiano Arce
Hospital de Basurto, Bilbao (Vizcaya).
Introducción: La Hemofilia A es la coagulopatía congénita más frecuente y grave en la infancia. Afecta a uno de cada 5000 varones a través de un patrón de herencia recesivo ligado al X. El momento del diagnóstico y la gravedad clínica está en correlación con la concentración en plasma del factor VIII de la coagulación. El 90% de las formas graves se presentan en el primer año de vida.
Caso clínico: Lactante varón de 4 meses que presenta intenso hematoma en extremidad superior izquierda. 24 horas antes se realiza una venopunción para estudio de Síndrome febril. Antecedentes: Padres jóvenes, sanos no consanguíneos. Hermana de 6 años, sana. Embarazo, parto y período neonatal sin incidencias. Exploración física: Afebril. TA:100/50. FC:140 lpm. Irritable. Gran tumefacción en extremidad superior izquierda con hematoma intenso en hueco antecubital y palmar de antebrazo izquierdo. No frialdad periférica. Limitación de la movilidad proximal. Pruebas complementarias: Hb: 8,6 g/dl. Hto: 24,4%. Plaquetas: 251.000/mm3. Leucocitos: 9.600/mm. Hemostasia: TTPA: 114,6 sg (25-40). APTT (mezcla): 42,9 sg. Indice de protrombina: 83%. INR: 1,14. Tiempo de trombina: 21sg. Fibrinógeno funcional: 210 mg %. Dímeros D: 0,4 mcg/ml. Recuento factorial: Factor VIII/C: < 1%. Factor von Willebrand, Factor IX/C, Factor XI/C y Factor XII/C: normales. Anticoagulante lúpico: negativo. Inhibidor factorial: negativo. Evolución y Tratamiento: Se instaura hemoterapia reposicional con plasma fresco y medidas locales disminuyendo el volumen del hematoma durante el período de estancia hospitalaria.
Discusión: Tras el estudio diagnóstico efectuado destaca el intenso retraso de formación de la protrombinasa endógena con corrección tras la adición de plasma control que, con una clínica hemorrágica asociada y la presencia de menos del 1% del Factor VIII indica una forma grave de Hemofilia A. Aunque en nuestro caso no hay evidencia de antecedentes familiares, hasta un 30% son esporádicos en relación a mutaciones de novo.
Conclusiones:1) La sospecha de un trastorno de la coagulación requiere reunir cuidadosamente antecedentes, datos clínicos y pruebas de laboratorio. 2) Las formas graves de Hemofilia A precisan un diagnóstico precoz y planificar el tratamiento para evitar complicaciones. 3) Es importante realizar un estudio genético para determinar la forma de herencia y localizar futuras portadoras.
P366 SÍNDROME HEMOFAGOCÍTICO SECUNDARIO EN PACIENTE CON LUPUS ERITEMATOSO SISTÉMICO. 13:35 h
Laura San Feliciano Martín, Manuela Muriel Ramos, Dorotea Raquel Fernández Álvarez, M. José Hernández Bejarano, Francisco José Fernández Pastor, Sonia de Arriba Méndez, Susana González de la Gándara, M. Dolores García García
Hospital Universitario, Salamanca.
El Síndrome Hemofagocítico es un desorden del sistema inmune caracterizado por la proliferación benigna de linfocitos T con activación exagerada de macrófagos y hemofagocitosis en bazo, médula ósea, ganglios, hígado, etc. Existe una forma primaria o familiar y formas secundarias relacionadas con infecciones víricas por VEB o CMV, infecciones bacterianas, micóticas, enfermedades autoinmunes tipo artritis reumatoide, neoplasias inmunodeficiencias, fármacos...
Nuestro objetivo es presentar un caso de asociación de S. Hemofagocítico con LES y resaltar la importancia del diagnóstico y las graves alteraciones que produce esta enfermedad, que son responsables de alta mortalidad, pero que pueden ser reversibles con un tratamiento temprano agresivo.
Caso clínico: Paciente varón de 13 años de edad diagnosticado de poliendocrinopatia autoinmune y recientemente de LES en tratamiento con prednisona a 2 mg/Kg/día. Tras 6 semanas de
