







El estudio del retraso mental hereditario, bajo el punto de vista diagnóstico y etiológico, es un gran reto. Una forma particular de retraso mental es el ligado al cromosoma X que se clasifica en formas sindrómicas y no sindrómicas, según la presencia o ausencia de un patrón físico, neurológico o metabólico específico asociado al retraso mental.
Pacientes y métodoSe han estudiado cinco generaciones de una familia con ocho hombres que padecía retraso mental. A seis de estos hombres se les ha estudiado clínicamente con medidas antropométricas e investigaciones genéticas: cariotipos de alta resolución, estudio molecular de X frágil, estudios de ligamiento y de los genes MID1 y PQBP1.
ResultadosEl estudio clínico mostró, junto a retraso mental, un patrón de microcefalia, micrognatia, anomalías osteoarticulares y genitales, talla baja y otras malformaciones menos frecuentes. Los cariotipos fueron normales y la investigación de mutaciones de los genes MID1 y PQBP1 fue negativa. El estudio de ligamiento mapeó el posible gen causal de este síndrome de retraso mental y anomalías congénitas múltiples en el segmento Xp11.23-q21.32, con un LOD score de 2.
ConclusionesHasta donde sabemos no está descrito un cuadro clínico como el que presentan estos enfermos que esté ligado a este segmento de X.
Sugerimos que esta familia padece un «nuevo síndrome» de retraso mental y anomalías congénitas múltiples ligado al cromosoma X.
Researching inherited mental retardation, from a diagnostic and aetiological point of view, is a great challenge. A particular type of mental retardation is the one linked to the X chromosome which is classified under syndromic and non-syndromic types, according to the presence or absence of a specific physical, neurological or metabolic pattern associated with mental retardation.
Patients and methodFive generations of a family have been studied with eight males suffering from mental retardation. Six of these males were clinically tested using anthropometric indicators and genetic tests: high resolution karyotypes, fragile X research, linkage and MID1 and PQBP1 gene studies.
ResultsAlong with mental retardation, the clinical study showed a pattern of microcephaly, micrognathia, osteoarticular and genital anomalies, short stature and other less frequent malformations. The linkage study mapped the possible causal gene of this mental retardation syndrome and multiple congenital abnormalities in the Xp11.23-q21.32 segment, with a LOD score of 2. As far as we know, a medical profile, similar to the one these patients have, linked to this X segment has not been described.
ConclusionsWe suspect that this family has a “new syndrome” of mental retardation and multiple congenital anomalies linked to the X chromosome.
Retraso mental es un síntoma caracterizado por un coeficiente intelectual (CI) significativamente por debajo del promedio (CI<70) junto con significativas limitaciones en la conducta adaptativa, que ocurre antes de los 18 años de edad1. Penrose2 observó un exceso de hombres entre los individuos con retraso mental y se explicó por la presencia de mutaciones en un gen ubicado en el cromosoma X3. Se calcula que el retraso mental ligado a X (RMLX) supone el 20-25% de los varones mentalmente retrasados4,5. Habitualmente el RMLX se clasifica en formas sindrómicas (RMXS) y no-sindrómicas (RMX), dependiendo de la presencia o ausencia de un patrón físico, neurológico o metabólico específico junto del retraso mental, aunque los límites entre un grupo y otro empiezan a desvanecerse desde que sabemos que, en ocasiones, mutaciones en un mismo gen son responsables de ambas formas.
Nosotros vamos a describir una familia en la que ocho de sus componentes masculinos estaban afectos de retraso mental y uno falleció de recién nacido. Hemos estudiado a los seis individuos vivos con retraso mental de esta familia en cuatro generaciones (fig. 1), pero aportamos fotos de otros dos fallecidos con anterioridad a nuestra investigación.
Pacientes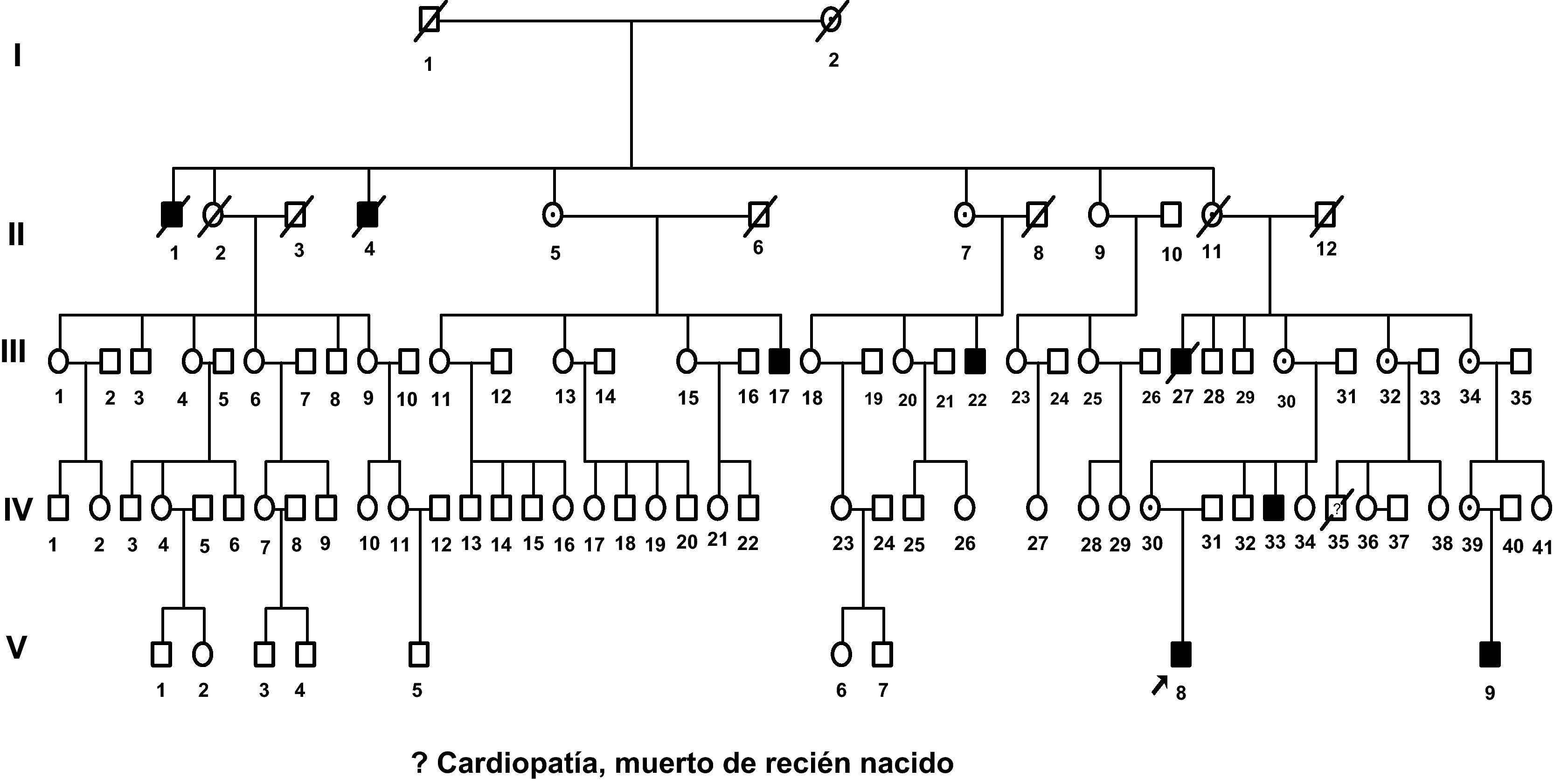
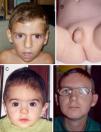
V-8 (fig. 2 A y B).
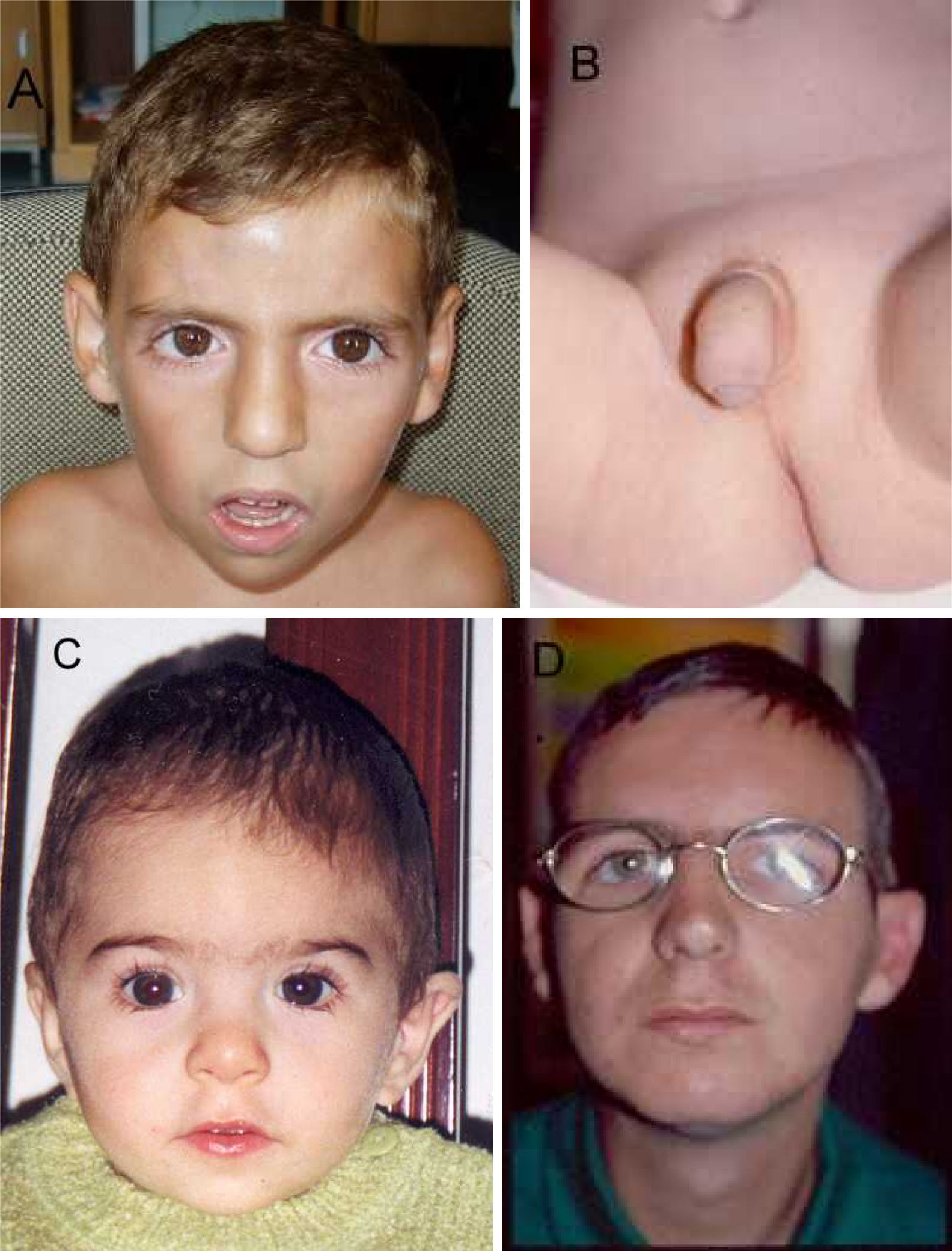
Trece años. Es hijo de padres jóvenes y sanos, Embarazo de 39 semanas con oligoamnios. Peso 2.700g. Talla 45cm (-3,8 DS). PC 32cm (-2,9 DS). Puntuación de Apgar 6-8. Fontanela 5×5cm, hipertelorismo, hendiduras palpebrales mongoloides. Surco subpalpebral marcado. Puente nasal ancho, premaxila protruida, hipertrofia gingival, paladar estrecho. Pene pequeño con prepucio hipoplásico que deja el glande al descubierto. Escroto hipoplásico y en bufanda, criptorquidia bilateral. Once pares de costillas. Estenosis aórtica leve y CIV.
Inició la deambulación a los 18 meses.
A los 4 años presentó crisis de ausencia con expresión EEG que no se han repetido con tratamiento específico durante tres años.
A los 6 años: pesaba 12.700g (-2,6 DS), su talla era 100cm (-3 DS) y su PC 48cm (-4,4 DS). Cara pequeña, bitemporal estrecho, hendiduras palpebrales grandes, ojos muy abiertos que originan una expresión de asustado, boca pequeña, paladar estrecho, micrognatia.
Come mal, se atraganta con frecuencia, náuseas de etrusión. Heces caprinas. No precisa tratamiento por su cardiopatía. Le intervinieron de orquidopexia y le colocaron una prótesis testicular.
Habla poco y mal. Es alegre, simpático, interactivo, tolera mal la frustración. CI 45.
V-9 (fig. 2 C).
Diez años. Nació a las 40 semanas de gestación, pesó 3.800g, midió 49cm, PC 33,5, puntuación de Apgar 7-8. Empezó a andar a los 18 meses.
A los tres años pesaba 10.500g (-2,6 DS), medía 91cm (-1,7 DS), PC 48cm (-2,4 DS). Hipertelorismo ocular, sinofridia. Boca pequeña, los premolares inferiores derechos amorfos. Micrognatia. Meñiques hipoplásicos, con discreta camptodactilia y sindactilia membranosa. Pene y escroto pequeños, pene incurvado, testes pequeños. Talones en mecedora. Hipotonía discreta, abdomen algo abombado. Empezó a hablar a los cuatro años. Es cariñoso, buen apetito, duerme bien.
Ácido láctico, pirúvico, ácidos orgánicos, cortisol, ACTH, aldosterona, fondo de ojo, Eco cerebral, potenciales evocados auditivos (PEA), resonancia magnética cerebral (RMC), ecocardiograma, EEG normales. Radiografía de cráneo: craneosinostosis coronal.
IV-33 (fig. 2 D).
31 años. Mide 163cm (-2DS), pesa 53kg (-1,5 DS), PC 53cm (-2,1 DS). Micrognatia. Hombros estrechos, cifosis discreta. Dedos índices con uñas hipoplásicas y flexión de las últimas falanges. Criptorquidia derecha, escroto hipoplásico. Pies planos, valgos. Se diagnosticó de pentalogía de Fallot y le intervinieron. Anduvo a los 18 meses. Estudio oftalmológico: resto de membrana pupilar en ojo derecho y adherencia del iris a la cara anterior del cristalino con opacidad. Mal rotación del riñón derecho. Tuvo un accidente vascular cerebral a los 26 años. Se atraganta frecuentemente. No presenta trastornos del lenguaje, es interactivo y se hace querer por el entorno. CI 50.
III-17 (fig. 3 A).
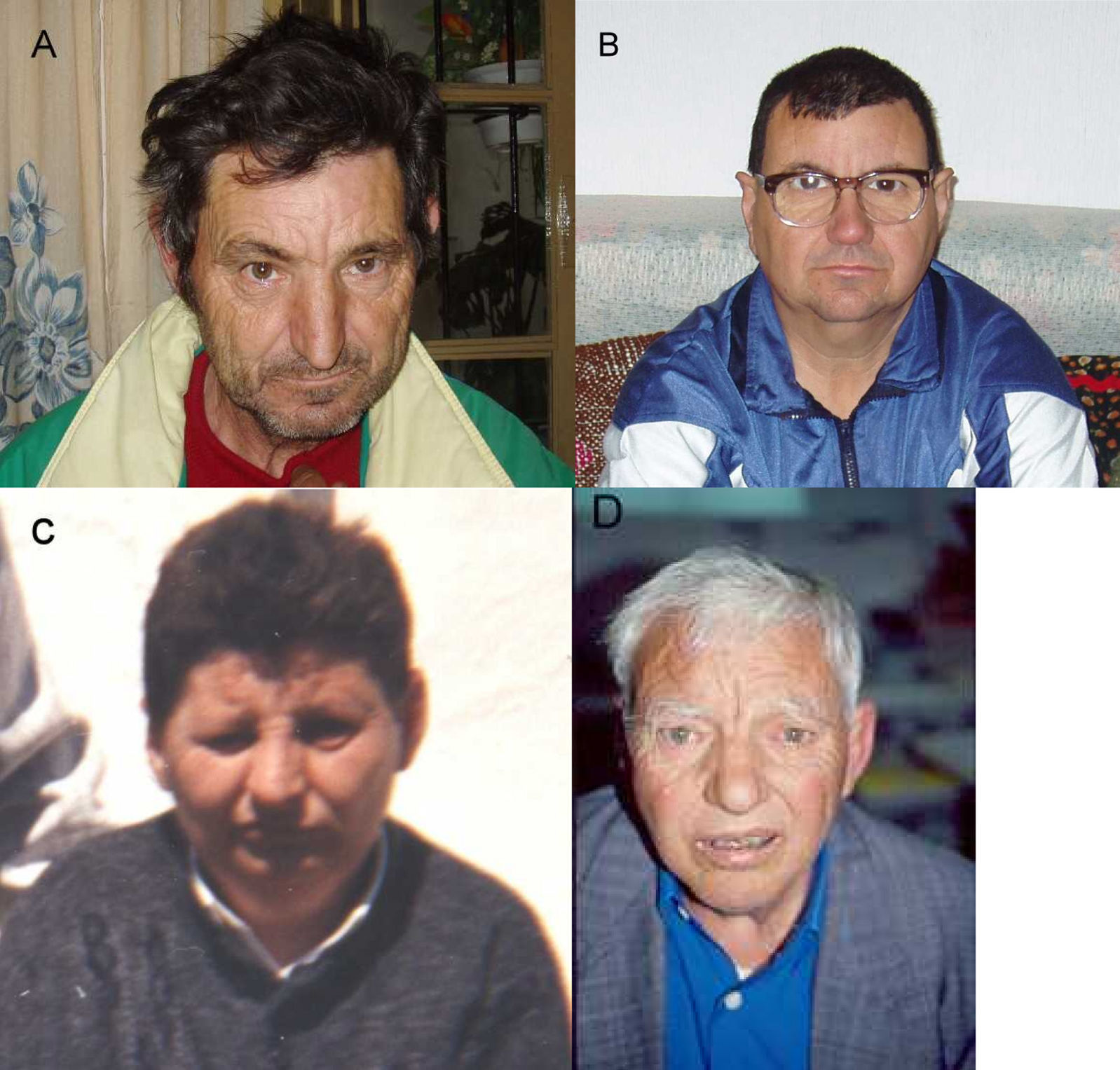
Talla161cm (-2,4 DS), peso 50kg (-1,8 DS), PC 50cm (-4 DS). Ojos mongoloides, nariz aguileña, filtro corto, labios finos, micrognatia. Hombros hacia delante, cierta cifosis. Mala tolerancia a la frustración, golpeándose o rompiendo cosas. CI 42.
III-22 (fig. 3 B).
Talla 168cm (-1 DS), peso 62kg (-0,4 DS), PC 51cm (-3 DS). Hombros hacia delante, cierta cifosis, criptorquidia. Carácter similar a III-17. CI 45.
III-27 (fig. 3 C).
Falleció a los 47 años. Por las fotografías se puede ver que tenía: microcefalia, hendiduras palpebrales mongoloides y cortas, nariz prominente, micrognatia. La familia nos comunica que tenía criptorquidia e hipospadias.
II-1 (fig. 4).

Falleció a los 42 años. Tenía RM. Por la foto podemos apreciar talla baja, microcefalia, nariz prominente y micrognatia.
II-4 (fig. 3 D).
Falleció a los 75 años. Talla 148cm (-4,5 DS), peso 39kg (-4 DS), PC 52cm (-2,7 DS), ojos mongoloides, micrognatia. Hablaba mal. CI 45.
Las mujeres, portadoras obligadas, no presentan ni RM ni malformaciones.
Investigaciones genéticasCon el consentimiento informado de todos los participantes o de los padres o hermanos en caso de ser menores de edad o incapacitados por su RM, se realizaron cariotipos de alta resolución (600 bandas), estudio molecular de síndrome de X frágil y estudio del gen PQBP1 por secuenciación de todo el gen a V-8, V-9, IV-33 y II-4. Estudio de MID1 al caso index. Estudio de ligamiento con microsatélites a los enfermos V-8, V-9, IV-33 y II-4, a las portadoras obligadas II-5, II-7, II-11, III-30, IV-30 y IV-39 y a los individuos III-11, III-13, III-15, III-28, III-29, III-32, IV-31, IV-32, III-34 y IV-40.
ResultadosEl cuadro clínico que describimos en esta familia se caracteriza (tabla 1) por retraso mental leve o moderado, microcefalia, micrognatia, anomalías genitales y osteoarticulares en el 100% de los casos y los siguientes signos en un porcentaje variable: talla baja, cara pequeña, nariz prominente, maxila prominente, boca pequeña, paladar estrecho, encías hipertróficas, cardiopatía congénita. Las anomalías genitales son variables: Pene pequeño, hipospadias y escroto hipoplásico, pero la criptorquidia es la más frecuente. Las malformaciones osteoarticulares son leves y aparecen en la cabeza, tronco, manos y pies.
Signos clínicos de los pacientes y su frecuencia
| Anomalías | V-8 | V-9 | IV-33 | III-17 | III-22 | III-27 | II-1 | II-4 | % |
| RM | + | + | + | + | + | + | + | + | 100 |
| Microcefalia | + | + | + | + | + | + | + | + | 100 |
| Micrognatia | + | + | + | + | + | + | + | + | 100 |
| Osteo-articular | + | + | + | + | + | D | D | + | 100 |
| Genitales | + | + | + | D | + | + | D | + | 100 |
| Oculares | + | + | + | + | − | + | D | + | 86 |
| Orales | + | + | + | + | − | − | D | − | 57 |
| Talla baja | + | +/− | − | + | − | − | + | + | 57 |
| Nasales | + | − | − | + | − | + | + | − | 50 |
| Cardiacas | + | − | + | − | − | − | − | − | 25 |
| Renales | − | − | + | − | − | − | − | − | 13 |
| CI | 45 | D | 50 | 42 | 45 | D | D | 39 |
D: desconocida.
La mayor parte de los rasgos fenotípicos que lo caracterizan asientan en la cabeza (craneales y faciales) y en los genitales.
Los cariotipos de alta resolución no mostraron anomalía. Las investigaciones para detectar mutaciones en los genes MID1 y PQBP1 fueron negativas. El estudio molecular de síndrome de X frágil fue normal. El estudio de ligamiento realizado a esta familia mapeó el posible gen causal de este síndrome de retraso mental y anomalías congénitas múltiples en el segmento Xp11.23-q21.32, con un LOD score de 2.
DiscusiónSe han descrito 22 síndromes de retraso mental ligado a X que mapean en Xp11.23-q21.32 (tabla 2), pero las diferencias fenotípicas con los pacientes aquí descritos son marcadas a nuestro entender, salvando algunos pacientes en los que se han demostrado mutaciones del gen PQBP1, los cuales comparten con los nuestros un buen número de malformaciones: Sutherland et al6 describieron una familia alguno de cuyos varones tenían retraso mental, talla baja, microcefalia, braquicefalia, diplejía espástica, testes pequeños y posiblemente CIR.
Síndromes de RMLX que mapean en Xp11.23-q21.32
| Síndrome | Rasgos principales | Gen afecto | OMIN | Ref |
| Síndrome de Renpenning: síndrome de Sutherland–Haan, síndrome de Hamel cerebro-palatocardiaco, síndrome de Golabi–Ito–Hall | Microcefalia, talla baja, hábito delgado, facies alargada, cardio-patía congénita, hendidura pala- tina, testes pequeños, diplejía espástica. | PQBP1 | 309500 | (12 6) |
| Síndrome FG 1 | Hipotonía congénita, ano imperforado, macrocefalia, agenesia parcial de cuerpo calloso. | MED12 | 305450 | (13) |
| Síndrome de Aarskog-Scott | Talla baja, hipertelorismo, escroto en bufanda, braquidactilia. | FGD1 | 305400 | (14) |
| Enfermedad de Menkes | Letargia y convulsiones desde el nacimiento. | MNK | 309400 | (15) |
| Síndrome de facies hipo- tónicas y RMLX: S. de Smith-Fineman-Myers, S. de Juberg-Marsidi, S. de Holmes-Gang, S. de Chudley-Lowry, S. de Carpenter-Waziri | Facies dismórficas e hipotónicas y en las mujeres portadoras un patrón de inactivación de X muy distorsionado. Otros síntomas: talla corta, sordera, hipogonadismo, anomalías renales y defectos esqueléticos leves. | ATRX | 309580 | (16) |
| Síndrome de Alfa-talasemia y RM | Similar al anterior y alfa-talasemia. | ATRX | 301040 | (17) |
| Síndrome de Siderius | Cara larga, punta de la nariz gruesa, labio leporino y hendidura palatina. | PHF8 | 300263 | (18) |
| Síndrome de Ahmad o MRXS7 | Obesidad, hipogonadismo y dedos adelgazados hacia la punta. | Desconocido | 300218 | (19) |
| Síndrome de Stocco Dos Santos | Luxación de caderas bilateral y talla baja. | SHROOM4 | 300434 | (20) |
| Síndrome de Prieto | Atrofia cerebral, dientes muy espaciados y en doble fila, nariz fina y prominente, mandíbula hipoplásica, atrofia papilar. | Desconocido | 309610 | (21) |
| Síndrome de Partington | Movimientos distónicos, disartria, convulsiones. | ARX | 309510 | (22) |
| Síndrome de Abidi | Talla baja, microcefalia, orejas grandes y en copa, hipotelorismo y testes pequeños. | Desconocido | 300262 | (23) |
| Síndrome de Miles-Carpenter | Microcefalia, cara asimétrica, ptosis, estrabismo, hendiduras palpebrales cortas, hipogonadismo, hipermotilidad articular, camptodactilia. | Desconocido | 309605 | (24) |
| Síndrome de Allan-Herndon-Dudley | Disartria, ataxia, movimientos atetoides, paraplejia espástica. Cara alargada, bitemporal estrecho, orejas grandes. | SLC16A2 | 300523 | (25) |
| Síndrome de Wieacker-Wolff | Contracturas de los pies, atrofia muscular distal progresiva y apraxia oculomotora. | Desconocido | 314580 | (26) |
| Síndrome de Brunner | Comportamiento agresivo episódico | MAOA | 300615 | (27) |
| RMXS relacionado con el gen JARID1C | Paraplejia espástica lentamente progresiva, hipotonía facial e hipoplasia maxilar, espectro autista. | JARID1 C | 300534 | (28) |
| Síndrome de Wilson-Turner | Obesidad, ginecomastia, pies pequeños dificultades del lenguaje. | Desconocido | 309585 | (29) |
| MRXS9 | Microcefalia, talla baja variable y lenguaje severamente retrasado. | Desconocido | 300709 | (30) |
| MRXS10 | Coreoatetosis, anormal comportamiento y aracnodactilia. | HSD17B10 | 300220 | (31) |
| RMXS tipo Turner | Macrocefalia, limitación de la extensión de los codos. | HUWE1 | 300706 | (32) |
| MRXS12 | Mutismo, fallo del crecimiento, convulsiones, braquicefalia, cara cuadrada, boca grande, labios gruesos y prognatismo. | Desconocido | 309545 | (33) |
OMIM: Online Mendelian Inheritance in Man; Ref:referencia, S.: síndrome.
Hamel et al7 publicaron una familia con RM severo, talla baja, cardiopatía grave, anomalías craneofaciales: hendidura palatina y paladar ojival, microcefalia, orejas anormales, nariz bulbosa, hipoplasia malar, boca pequeña y micrognatia. Espasticidad y muerte precoz.
Degaqi et al8 publicaron otra familia que interpretaron como RMX (MRX55).
Kalscheuer et al9 publicaron una familia parecida a la de Sutherland pero sin diplejía espástica y sin testes pequeños. Otra familia tenía RM, microcefalia, atresia anal y situs inverso y comprobaron que los pacientes afectos tenían una mutación en el gen PQBP1 ubicado en Xp11.23.
Posteriormente se han descrito otras siete familias con mutaciones en PQBP1, incluyendo la publicada por Martínez-Garay et al10 con retraso mental y diplejía espástica.
Stevenson et al11 revisaron los signos clínicos de los individuos afectados de diez familias a los que se les había encontrado una mutación en el gen PQBP1 y concluyeron que los más comunes son retraso mental, microcefalia, talla baja y testes pequeños. Propuso que a todos ellos se les denominara síndrome de Renpenning12.
Nuestros enfermos tienen en común con los publicados bajo este epígrafe el retraso mental, microcefalia, micrognatia, talla baja, testes pequeños, algunas anomalías faciales y en algún caso cardiopatía, pero no espasticidad ni diplejía.
El síndrome de Renpenning es paradigmático de la variabilidad fenotípica a que dan lugar en algunas ocasiones las anomalías genéticas producidas en un único gen, lo cual nos sugiere que la interacción entre los productos de los genes puede ser la responsable de esta variabilidad.
El síndrome de facies hipotónicas y RMLX16 es producido por mutaciones del gen ATRX ubicado en Xq13. Este término comprende varios síndromes descritos por separado: Smith-Fineman-Myers, Juberg-Marsidi, Holmes-Gang, Chudley-Lowry y Carpenter-Waziri.
El síndrome de alfa-talasemia y RM17 es alélico al anterior, tiene un fenotipo parecido, pero asociado a alfa talasemia: hipocromía, microcitosis y la existencia de cuerpos de Hb H. Ninguno de nuestros pacientes presentó alfa-talasemia.
Destacamos la importancia de nuestros hallazgos desde un punto de vista práctico, por un lado, para el asesoramiento genético de esta familia y desde una perspectiva científica, para la investigación e identificación de un nuevo gen implicado en un nuevo síndrome de RMLX. La aplicación de las nuevas técnicas moleculares actualmente disponibles, como microarrays o secuenciación masiva, podrán determinar en un futuro cercano la mutación genética causal específica de este síndrome. Su identificación permitiría a su vez la detección de mujeres portadoras en riesgo de tener hijos afectos de RMLX y su correcto asesoramiento genético, propiciando a partir de ese momento el potencial uso de técnicas de diagnóstico genético prenatal y/o preimplantacional específico.
En resumen, Describimos un nuevo síndrome de RMLX34 caracterizado por microcefalia, micrognatia, malformaciones osteoarticulares leves y anomalías genitales que mapea en Xp11.23-q21.32.
De forma inconstante se asocian talla baja y otras anomalías faciales o en otras regiones anatómicas.
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.
Estamos muy agradecidos a los enfermos y a sus familias por su colaboración. Al Dr. Antonio Pérez Aytés por su ayuda con el texto. A los Dres. Germana Meroni, Rosa Ferrentino, Laura Esposito y Massimo Carella por los estudios de ligamiento y del gen MID1. A la Dra. Vera Kalscheuer por el estudio del gen PQBP1.
