
El síndrome de Cornelia de Lange (SCdL) es una enfermedad congénita rara con afectación multisistémica y del desarrollo, que abarca un gran espectro fenotípico1–3. El fenotipo clásico se caracteriza por rasgos craneofaciales distintivos (sinofridia, cejas gruesas y arqueadas, puente nasal corto, narinas antevertidas y microcefalia), retraso del crecimiento de comienzo prenatal, discapacidad intelectual, hipertricosis y defectos por reducción de las extremidades1,2. El fenotipo no clásico tiende a ser más leve.
El 60% de las personas afectadas presentan variantes patogénicas en el gen NIPBL (herencia autosómica dominante). Otros genes implicados son RAD21, SMC3 y BRD4 (herencia autosómica dominante) y HDAC8 y SMC1A (herencia ligada a X). En la mayoría de los pacientes (>95%) las variantes patogénicas ocurren de novo. El mecanismo fisiopatológico es la haploinsuficiencia3,4.
Las guías del American College of Medical Genetics and Genomics y de la Association for Molecular Pathology determinan que las variantes clasificadas como «patogénicas» y como «probablemente patogénicas» son diagnósticas de este síndrome, no así las de «significado incierto».
Los genes implicados se encuentran asociados al complejo de cohesinas1,4. Este complejo es multifuncional y participa en numerosos procesos biológicos, entre los que destaca la regulación de la expresión génica4, por lo que un fallo en el funcionamiento de este complejo produce una desregulación global de la expresión génica, lo que explica el gran espectro fenotípico y la afectación multisistémica de este síndrome.
Exponemos el caso de una niña de 8 años con SCdL que presenta una variante patogénica en el gen NIPBL en heterocigosis no descrita previamente.
Se trata de una niña en seguimiento en consulta de Endocrinología Infantil por pequeña para la edad gestacional simétrica sin crecimiento recuperador, talla baja (−2,73DE) con edad ósea retrasada, hipertricosis y fenotipo peculiar (sinofridia, epicantus, narinas antevertidas, micrognatia y microcefalia). Presenta función hipofisaria y función tiroidea conservadas y desarrollo uterino normal sin alteraciones genitales. Asimismo, presenta clinodactilia del quinto dedo de ambas manos. Además, se encuentra en seguimiento multidisciplinar por bajo peso, discapacidad intelectual leve, trastorno del lenguaje y microcefalia.
Ante el cuadro sindrómico indicativo de SCdL con 9 puntos en el score clínico de Kline5, en 2018 se deriva a la paciente al Servicio de Genética de nuestro centro de referencia para estudio. Se realiza hibridación genómica comparativa a base de microarrays (aCGH) y secuenciación dirigida del exoma, siendo el resultado negativo para este síndrome. No se detectan tampoco otras alteraciones que justifiquen el cuadro clínico.
No obstante, dada la alta sospecha clínica, en 2023 se solicita ampliación del estudio genético en el laboratorio externo de referencia qGenomics. Se realiza una secuenciación masiva del exoma, detectándose la variante c.3855+4A>G heterocigota de tipo splicing en el gen NIPBL. Se concluye que esta variante afecta al sitio donador de splicing del exón 16, y aunque no afecta al sitio canónico de splicing, podría alterar el procesamiento del ARN mensajero y dar lugar a una proteína aberrante. Asimismo, la variante no se ha observado en individuos de la base de datos de variantes comunes ni se ha identificado en pacientes hasta la fecha, por lo que catalogan la variante como de «significado incierto» asociada al SCdL tipo 1.
Por otra parte, el estudio de segregación en muestras parentales ha demostrado que la variante se originó de novo.
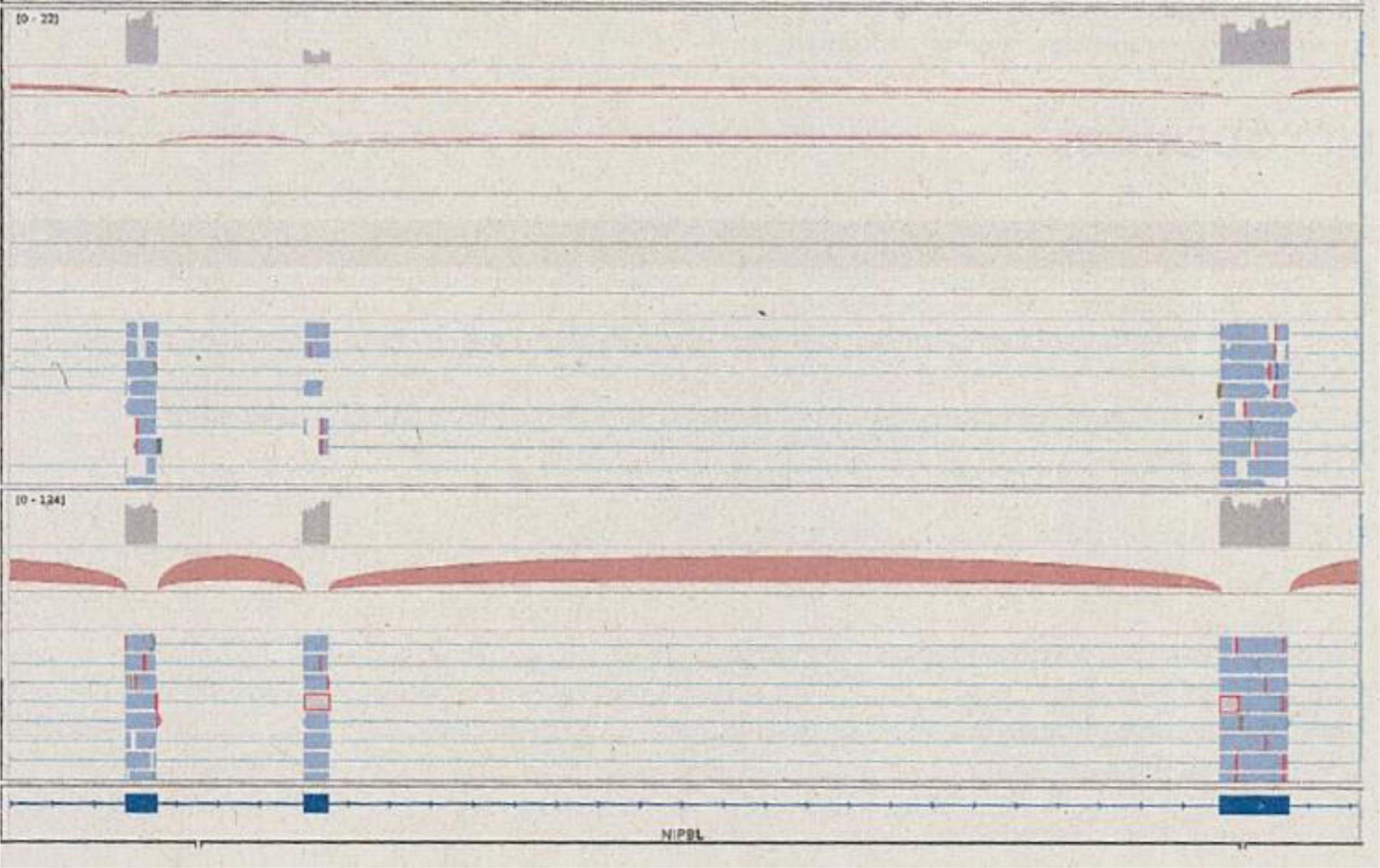
Ante una variante de «significado incierto» se amplía el análisis con estudios funcionales. Se realiza secuenciación del transcriptoma (RNAseq), detectándose que la presencia de la variante descrita no altera los niveles de expresión del gen NIPBL, pero sí se observa un efecto sobre la estructura del transcrito producido, eliminándose el exón 16 (exón skipping), sin alterarse la pauta de lectura (fig. 1). Concluyen que es posible predecir que esta alteración causaría una haploinsuficiencia funcional, compatible con el mecanismo fisiopatológico conocido para el SCdL asociado a NIPBL. Por ello, esta información permite reclasificar la variante como «patogénica» según las guías del American College of Medical Genetics and Genomics y de la Association for Molecular Pathology, y diagnosticar a nuestra paciente de SCdL tipo 1.

Lecturas que cubren los exones 15, 16 y 17. En la parte superior se observa una combinación de lecturas que soportan la presencia del transcripto habitual, junto con otras que excluyen el exón 16 y empalman el 15 con el 17. En la parte inferior, se presentan resultados de un individuo control, donde solo se observan lecturas que soportan la presencia del transcrito de los 3 exones. Fuente: Extraída del informe de qGenomics.
Lo relevante del caso es que la realización de la técnica de secuenciación del transcriptoma (RNAseq) ha permitido reclasificar una variante de «significado incierto» en una variante «patogénica», pudiendo así diagnosticar a nuestra paciente de SCdL tipo 1, así como añadirla como nueva variante patogénica de este síndrome en las bases de datos genéticas internacionales.

