



La histiocitosis de células de Langerhans (HCL) es actualmente considerada una neoplasia inflamatoria rara de origen mieloide que afecta a diferentes órganos y sistemas, de presentación clínica heterogénea. La HCL neurodegenerativa (HCL-ND) es una entidad tardía y poco frecuente, caracterizada por un cuadro clínico-radiológico progresivo que supone una importante morbilidad, con una evolución variable e impredecible. Históricamente fue considerada una secuela de probable etiología paraneoplásica, con tratamientos poco efectivos. La afectación multisistémica, la presencia de diabetes insípida central y la afectación ósea orbitaria y/o de la base de cráneo se consideran factores de riesgo asociados al desarrollo de la HCL-ND. El diagnóstico precoz, eminentemente clínico y radiológico, apoyado por estudios electrofisiológicos y neurocognitivos, permitiría un tratamiento temprano, posiblemente clave para revertir o ralentizar la evolución. En los últimos años, el descubrimiento de la presencia de mutaciones activadoras de la vía dependiente de la proteína quinasa de activación mitogénica (MAPK) y el desarrollo de modelos animales en ratón han permitido redefinir esta entidad como una forma de enfermedad activa de HCL que produce neuroinflamación y neurodegeneración, suponiendo un cambio en el conocimiento de la etiopatogenia, identificando nuevos factores de riesgo y posibles tratamientos dirigidos.
Langerhans cell histiocytosis (LCH) is a rare inflammatory myeloid neoplasm with heterogeneous organ and system involvement. Neurodegenerative LCH (ND-LDH) is an uncommon late manifestation with progressive clinical and radiological features, substantial morbidity and an unpredictable course. Historically it was considered a paraneoplastic sequela for which treatment was largely ineffective. Multisystemic disease, central diabetes and orbital and/or skull base bone lesions are risk factors associated with the development of ND-LCH. Early diagnosis, guided by clinical, radiological, electrophysiological and neurocognitive evaluations, may enable timely treatment to halt or slow its progression. Recent identification of activating mutations in the mitogen-activated protein kinase (MAPK) pathway and the development of murine models have redefined ND-LCH as an active neuroinflammatory and neurodegenerative form of LCH, changing our understanding of its etiopathogenesis and identifying novel risk factors and targets for therapy.

La histiocitosis de células de Langerhans (HCL) es una enfermedad rara hematológica de origen mieloide que forma parte de las enfermedades histiocitarias y que afecta principalmente a la población pediátrica1,2. Tras largos años de debate sobre el origen neoplásico o inflamatorio de la enfermedad, la proliferación clonal3 y el descubrimiento de la mutación BRAFV600E (sustitución de valina por ácido glutámico en el aminoácido 600 del proto-oncogén B-Raf)4 u otras mutaciones activadoras de la vía dependiente de la proteína quinasa de activación mitogénica (MAPK), mutuamente excluyentes, como causa patogénica1, han permitido que sea finalmente considerada una neoplasia hematológica de origen mieloide por la OMS5.
La HCL presenta una gran heterogeneidad clínica, actualmente entendida gracias a los avances en el conocimiento de su biología: desde lesiones auto-involutivas a cuadros multiorgánicos graves que precisan tratamiento intensivo. La afectación ósea y mucocutánea son las más frecuentes. El compromiso de órganos de riesgo (hígado, bazo y médula ósea) confiere un peor pronóstico, con mayores tasas de recurrencia y peor supervivencia6. A pesar de los avances en el tratamiento, consiguiendo una excelente supervivencia7, las recaídas/reactivaciones siguen siendo un problema, incluso con el tratamiento dirigido, responsables de la morbilidad a largo plazo, sobre todo a nivel ortopédico y endocrinológico.
El sistema nervioso central (SNC) también puede verse afectado, pero dada su rareza y definición vaga y diversa, su incidencia y prevalencia reales no están del todo aclaradas en la literatura; se estima que hasta un 5-10% de los pacientes pueden desarrollar afectación del SNC8, en dos formas bien diferenciadas por su origen etiopatogénico y su patrón clínico-radiológico.
En casi la mitad de los casos se produce en forma de lesiones ocupantes de espacio, en localizaciones como la región hipotálamo-hipofisaria, los plexos coroideos, o lesiones con implantación dural. Estas lesiones están formadas por histiocitos con el patrón histológico clásico de las lesiones extracraneales (CD1a+/CD207+). De especial interés son los pacientes que desarrollan diabetes insípida central (DIC), ocurriendo en aproximadamente el 12% de los casos, y presente ya al momento del diagnóstico de la HCL en la mitad de los pacientes9-11. La afectación multisistémica u ósea craneofacial y un mayor tiempo de enfermedad activa o número de recurrencias son factores de riesgo de desarrollo de DIC (Hazard ratios de 4,6, 1,7, 1,5 y 3,5, respectivamente)9. Aunque existen casos de resolución de la DIC descritos en la literatura10, en la práctica totalidad de los pacientes permanece como secuela, y hasta un 40% pueden asociar otros déficits hipotálamo-hipofisarios8,12.
La segunda forma de afectación del SNC es la forma neurodegenerativa (ND), en la cual profundizaremos en este trabajo revisando los avances sobre el conocimiento de su etiopatogenia, el diagnóstico, y sus implicaciones en el tratamiento.
Enfermedad neurodegenerativa de la HCLAunque la supervivencia global de la HCL es alta6, un pequeño porcentaje de los pacientes desarrolla un síndrome progresivo de alteraciones radiológicas y manifestaciones clínicas neurológicas. En 2018 Héritier et al., en la mayor cohorte publicada de pacientes con HCL, describieron que hasta el 1,9% de los pacientes desarrollaban una enfermedad ND clínica, definida como exploración física alterada y/o disfunción neuropsicológica con patrón radiológico compatible. La incidencia acumulada a los 15años era del 1,8-8,6%, posiblemente subestimada, ya que pacientes paucisintomáticos podrían no haberse incluido. La media de edad al diagnóstico de HCL-ND era de 9,2años y ocurría habitualmente años después del diagnóstico de HCL (media de 6,5años)11.
Avances en la etiopatogenia de la enfermedadHasta el descubrimiento de la mutación BRAFV600E en el año 2010 como alteración molecular clave para el desarrollo de la HCL, se desconocía el origen de la enfermedad, y mucho menos se podía explicar su gran variabilidad clínica4. Actualmente sabemos que mutaciones activadoras de la vía MAPK en células de origen mieloide en diferentes momentos de su diferenciación reproducen las diferentes formas clínicas: cuanto más indiferenciadas, más agresivas. Esta activación patológica de la vía MAPK favorece la diferenciación hacia células con alteración de la migración (pérdida de CCR7), resistencia a la apoptosis (aumento de Bcl-xL) e inducción de un programa de senescencia celular, y produce un agotamiento de las célulasT circundantes por inhibición de los puntos de control inmune13.
En los primeros estudios de las escasas biopsias y autopsias del SNC se observaba destrucción neuronal y axonal con una infiltración difusa inflamatoria formada mayoritariamente por linfocitos CD8+ y microglía CD68+, pero CD1a−, excluyendo por tanto que fueran células derivadas de las células de Langerhans conocidas y, por tanto, no encontrado enfermedad activa. Por ello, la HCL-ND se describía como una secuela inmunomediada, paraneoplásica14.
Tras el descubrimiento molecular, McClain et al. revisaron a nivel histológico la enfermedad y demostraron la presencia de la mutación BRAFV600E en células del SNC con patrón perivascular difuso y fenotipo mielo-monocítico (CD14+/CD33+/CD163+/P2RY12-), lo cual suponía un cambio en la etiopatogenia, confirmando la presencia de células tumorales y, por tanto, enfermedad activa15. Este grupo de investigación, con datos clínicos y su modelo animal en ratón, defiende como origen de esta entidad células madre hematopoyéticas circulantes que, con la mutación BRAFV600E, son capaces de penetrar y proliferar en el SNC, ayudadas por una barrera hematoencefálica también alterada por el estado proinflamatorio producido por las mismas15,16. El modelo animal reproduce clínicamente la enfermedad con una afectación sistémica inicial y posterior neurodegeneración. Recientemente, Abagnale et al., con su modelo in vitro, apoyarían esta teoría17. Por otro lado, Mass et al. demostraron con otro modelo en ratón que BRAFV600E en progenitores mielo-eritroides tempranos, precursores de los futuros macrófagos residentes del SNC —la microglía—, reproducían también una enfermedad neuroinflamatoria y neurodegenerativa con respuesta a los inhibidores de MAPK (iMAPK), pero sin la afectación sistémica extra-SNC previa que la mayoría de los pacientes con HCL presentan18. Dicho grupo describe la HCL-ND como una enfermedad neuroinflamatoria clonal de la microglía asociada a la mutación BRAFV600E y plantea la posibilidad de que la HCL-ND pueda tener un doble origen: la microglía o células progenitoras procedentes de la médula ósea (fig. 1). La presencia de sintomatología dependería del tiempo de enfermedad activa y la cantidad de células clonales produciendo el daño a nivel del SNC19. Esta última idea converge con la reciente demostración de que la severidad de la afectación hematológica está directamente relacionada con la cantidad de mutación de BRAFV600E detectada en ADN libre en sangre periférica7.
Esquema de la vía de señalización dependiente de la proteína quinasa de activación mitogénica (MAPK). La unión de factores de crecimiento/mitógenos a los receptores tirosín quinasa produce la activación de Ras mediante su fosforilación. Esto produce una activación y homodimerización de Rafs (B-RAF o A-RAF) y la subsecuente señalización posterior de MAPK. La mutación BRAFV600E produce una activación de BRAF en forma de monómero, independientemente de Ras, favoreciendo una activación constitutiva de la vía MAPK. B)Los estudios clínicos y los modelos animales en ratón proponen dos posibles orígenes de la histiocitosis de células de Langerhans neurodegenerativa (HCL-ND). En el primero, una población clonal de la microglía residente en el sistema nervioso central (SNC) con mutaciones activadoras de la vía MAPK conllevaría a una proliferación de dicha población (microgliosis) productora de sustancias pro-inflamatorias. Dicha situación activaría los astrocitos de forma reactiva y neurotóxica, generando finalmente una pérdida neuronal. El origen embriológico de esta microglía activada serían progenitores mielo-eritroides tempranos, procedentes del saco vitelino, que en la hematopoyesis primitiva se escinden de forma muy precoz en el desarrollo. En un segundo modelo, células mieloides circulantes con mutación activadora de MAPK, procedentes de células madre hematopoyéticas de la médula ósea, llegarían al SNC, acumulándose y ocasionando el daño neuroinflamatorio. Asimismo, estas células producirían un estado pro-inflamatorio que alteraría la barrera hematoencefálica, hecho que facilitaría la entrada al SNC de esta población clonal. La probabilidad de desarrollo de sintomatología neurológica dependería del tamaño de estas poblaciones clonales situadas en el SNC y del tiempo de evolución.")
Modelo etiopatogénico de la HCL-ND.
A)Esquema de la vía de señalización dependiente de la proteína quinasa de activación mitogénica (MAPK). La unión de factores de crecimiento/mitógenos a los receptores tirosín quinasa produce la activación de Ras mediante su fosforilación. Esto produce una activación y homodimerización de Rafs (B-RAF o A-RAF) y la subsecuente señalización posterior de MAPK. La mutación BRAFV600E produce una activación de BRAF en forma de monómero, independientemente de Ras, favoreciendo una activación constitutiva de la vía MAPK. B)Los estudios clínicos y los modelos animales en ratón proponen dos posibles orígenes de la histiocitosis de células de Langerhans neurodegenerativa (HCL-ND). En el primero, una población clonal de la microglía residente en el sistema nervioso central (SNC) con mutaciones activadoras de la vía MAPK conllevaría a una proliferación de dicha población (microgliosis) productora de sustancias pro-inflamatorias. Dicha situación activaría los astrocitos de forma reactiva y neurotóxica, generando finalmente una pérdida neuronal. El origen embriológico de esta microglía activada serían progenitores mielo-eritroides tempranos, procedentes del saco vitelino, que en la hematopoyesis primitiva se escinden de forma muy precoz en el desarrollo. En un segundo modelo, células mieloides circulantes con mutación activadora de MAPK, procedentes de células madre hematopoyéticas de la médula ósea, llegarían al SNC, acumulándose y ocasionando el daño neuroinflamatorio. Asimismo, estas células producirían un estado pro-inflamatorio que alteraría la barrera hematoencefálica, hecho que facilitaría la entrada al SNC de esta población clonal. La probabilidad de desarrollo de sintomatología neurológica dependería del tamaño de estas poblaciones clonales situadas en el SNC y del tiempo de evolución.
Hasta la incorporación del diagnóstico molecular, los estudios clínicos describían la presencia de DIC como el mayor factor de riesgo de HCL-ND. Hasta un 70-90% de los pacientes con HCL-ND tienen DIC20-22. Los pacientes con HCL con mayor riesgo de DIC eran aquellos con afectación multisistémica o de los huesos de la base de cráneo o la órbita9; por ello, a dicha afectación ósea se la denominó como «lesiones de riesgo para el SNC».
Héritier et al. encontraron que la mutación BRAFV600E estaba estadísticamente relacionada con el desarrollo de DIC y la enfermedad ND, en comparación con los pacientes sin la mutación (19,8% vs. 8,4% [p=0,006] y 6,4% vs. 1,4% [p=0,04], respectivamente)23. Dicha mutación estaba también asociada a un diagnóstico en edades precoces, formas multisistémicas, afectación de piel y órganos de riesgo, refractariedad al tratamiento de primera línea y reactivaciones de la enfermedad, algunas de ellas descritas previamente como factores de riesgo de DIC9. En su estudio, combinando datos clínicos y moleculares, observaron que en aquellos pacientes «susceptibles de ND clínica» (definidos por haber tenido afectación ósea de base de cráneo/orbitaria y/o DIC), el riesgo a los 10años de HCL-ND clínica de los BRAFV600E era del 33,1%, comparado con el 2,9% de los pacientes sin la mutación (p=0,002)11.
Los tratamientos sistémicos convencionales utilizados históricamente para la HCL no parece que hayan tenido un impacto en la prevención de la HCL-ND. Además, con la generalización del uso de los iMAPK en formas refractarias o recaídas fuera del SNC, existe una cierta preocupación por cómo este tratamiento puede cambiar la historia de la enfermedad a largo plazo: algunos grupos han reportado un aumento de incidencia de alteraciones clínico-radiológicas de HCL-ND, y de forma más precoz, en pacientes tratados con estos inhibidores en monoterapia24. La combinación de quimioterapia con iMAPK controlaría la enfermedad sistémica, sin empeorar la supervivencia derivada por la toxicidad del tratamiento y evitando este aumento detectado de ND con la monoterapia25. Se precisa una mayor experiencia y un esfuerzo colaborativo para poder obtener respuestas a estas preguntas que aún se plantean.
Asimismo, en la era de la biología molecular, la detección BRAFV600E en sangre periférica al diagnóstico de la HCL podría convertirse en un marcador útil en el futuro, no solo para el diagnóstico y evaluación de la respuesta al tratamiento, sino también para predecir el desarrollo de la HCL-ND23,26.
Diagnóstico de la enfermedadLa HCL-ND es un síndrome progresivo de alteraciones radiológicas y manifestaciones clínicas. Algunos autores proponen definirlos separadamente, dada la mala correlación clínico-radiológica, y hablar de «anomalías radiológicas asociadas a HCL» (LCH-associated abnormal CNS imaging [LACI]) y «síntomas de afectación del SNC asociados a HCL» (LCH-associated abnormal CNS symptoms [LACS])27.
Sintomatología neurológica y neurocognitivaClínicamente se caracteriza por una afectación variable y progresiva de las funciones cerebelosas (temblor, ataxia, dismetría, disartria) y del tronco del encéfalo (disfagia, paresia de pares craneales). Los casos avanzados pueden desarrollar tetraparesia espástica y parálisis pseudobulbar. Existen varias escalas para cuantificar el grado de afectación neurológica. La Expanded Disability Status Scale [EDSS]), muy utilizada en esclerosis múltiple y aplicada en estudios de HCL, ofrece una cuantificación objetiva y reproducible de la discapacidad motora. Para la disfunción cerebelosa existen dos escalas que pueden ayudar al diagnóstico y seguimiento: la International Cooperative Ataxia Rating Scale (ICARS), con 19 ítems y puntuación máxima de 100, y la Scale for the Assessment and Rating of Ataxia (SARA), más sencilla, con 8 ítems y puntuación máxima de 40. La colaboración de un experto en neurología puede ser de ayuda en la aplicación de estas escalas. El diagnóstico clínico de HCL-ND por la Histiocyte Society se establece con una puntuación ICARS ≥5.
Existen pocos estudios que hayan investigado las alteraciones neuropsicológicas y cognitivas en pacientes con HCL28-30. Aquellos con afectación ND presentan un síndrome dis-ejecutivo que impacta en la esfera verbal, la memoria de trabajo, la atención y la velocidad de procesamiento. No obstante, la utilidad clínica de una evaluación basal y seriada aún no ha sido analizada, aunque podría contribuir a un diagnóstico y tratamiento tempranos, favoreciendo así la adaptación social y escolar. Por último, las alteraciones del humor y del comportamiento, poco estudiadas hasta el momento, han sido reportadas en hasta un 25% de los casos28.
Estudios de imagen en la HCL-NDLa resonancia magnética (RM) convencional es la herramienta de imagen más utilizada, aunque sus hallazgos son poco específicos. En el diagnóstico diferencial se incluyen entidades como la encefalomielitis aguda diseminada, enfermedades metabólicas y neurodegenerativas, leucoencefalopatía post-quimioterapia/radioterapia o encefalitis paraneoplásica.
Aunque determinar la incidencia real de las LACI en HCL es complejo, se estima que un 20-26% de los pacientes presentarían anomalías en la RM compatibles con HCL-ND en su evolución7,22,31. Dicha prevalencia se podría incrementar con la mejoría en la supervivencia de los pacientes de alto riesgo y los tratamientos dirigidos. Aun así, no existen en la actualidad recomendaciones claras sobre el seguimiento por imagen y su duración (fig. 2).

Estas alteraciones son predominantemente infratentoriales, con anomalías simétricas en los núcleos dentados y la sustancia blanca cerebelosa, hiperintensas en T2 y FLAIR. En fases más avanzadas, estas alteraciones pueden extenderse a los pedúnculos cerebelosos, la protuberancia y al bulbo raquídeo. También se han observado afectación de los ganglios basales (hiperintensidad en T1, intensidad variable en T2) y lesiones parcheadas en la sustancia blanca supratentorial, sin un patrón vascular claro (hiperintensidad en T2, hipointensidad en T1). Alteraciones tardías incluyen la atrofia cerebelosa, mesencefálica o del cuerpo calloso, y la dilatación de los espacios de Virchow, reflejo de la pérdida neuronal y axonal. La correlación clínico-radiológica es limitada, aunque hasta un 25% de los pacientes con alteraciones en la RM pueden desarrollar sintomatología con el tiempo11,27,31-33.
Los estudios de imagen funcional avanzada podrían ofrecer mayor sensibilidad y especificidad. Técnicas como la estimación de contenido de mielina o de densidad axonal, la espectroscopia (ratio N-acetil-aspartato/creatina), imágenes con difussion tensor imaging (DTI) y estudios con radioisótopos (F18-FDG PET) han sido exploradas, aunque aún se requieren más estudios para definir su papel en el diagnóstico y seguimiento34,35.
Evaluación neurofisiológicaLos estudios electrofisiológicos, aunque con experiencia limitada, pueden ser útiles para el diagnóstico, el seguimiento y la evaluación del tratamiento de la HCL-ND. Se han analizado tanto los potenciales evocados somatosensoriales (PESS) como los auditivos evocados de tronco (PEATC). Los PESS podrían desempeñar un papel importante en la detección precoz de la HCL-ND34, además de servir como posible marcador de respuesta al tratamiento35.
Marcadores bioquímicosActualmente no existen recomendaciones específicas para el análisis del líquido cefalorraquídeo (LCR) en pacientes con sospecha de HCL-ND. En el ámbito de la investigación se han explorado diversos biomarcadores que podrían ser útiles en el diagnóstico y el seguimiento de estos pacientes. Entre ellos, la osteopontina15 y el neurofilamento-L36, marcadores de inflamación y de daño axonal, respectivamente, han sido evaluados tanto en sangre periférica como en LCR y podrían tener un impacto como marcadores diagnósticos y de respuesta al tratamiento; sin embargo, se requieren más estudios prospectivos que ayuden a definir su papel en este contexto.
Papel de la biopsia de SNCLa biopsia cerebral se recomienda únicamente en casos donde el diagnóstico previo de HCL no haya sido confirmado y una biopsia en otra localización no sea posible. La demostración de la activación de la vía de MAPK (detectando la mutación BRAF por inmunohistoquímica o biología molecular, o de pERK por inmunohistoquímica) podría no solo establecer el diagnóstico, sino también identificar una posible diana terapéutica, lo que justificaría la realización de la biopsia. Los avances en la neurocirugía permiten en la actualidad realizar biopsias en localizaciones complejas con nula o muy baja morbilidad en centros de referencia con la experiencia y la tecnología adecuadas. Los marcadores de origen mieloide de la microglía (CD163, CD68) son positivos, pero la tinción clásica de HCL, CD1a, será negativa14,15. Por otro lado, la detección de BRAFV600E en pacientes con diagnóstico radiológico y/o clínico de HCL-ND ha sido documentada tanto en células mononucleares en sangre periférica (PBMC) como en LCR, la cual podría también emplearse como una forma de biopsia líquida15.
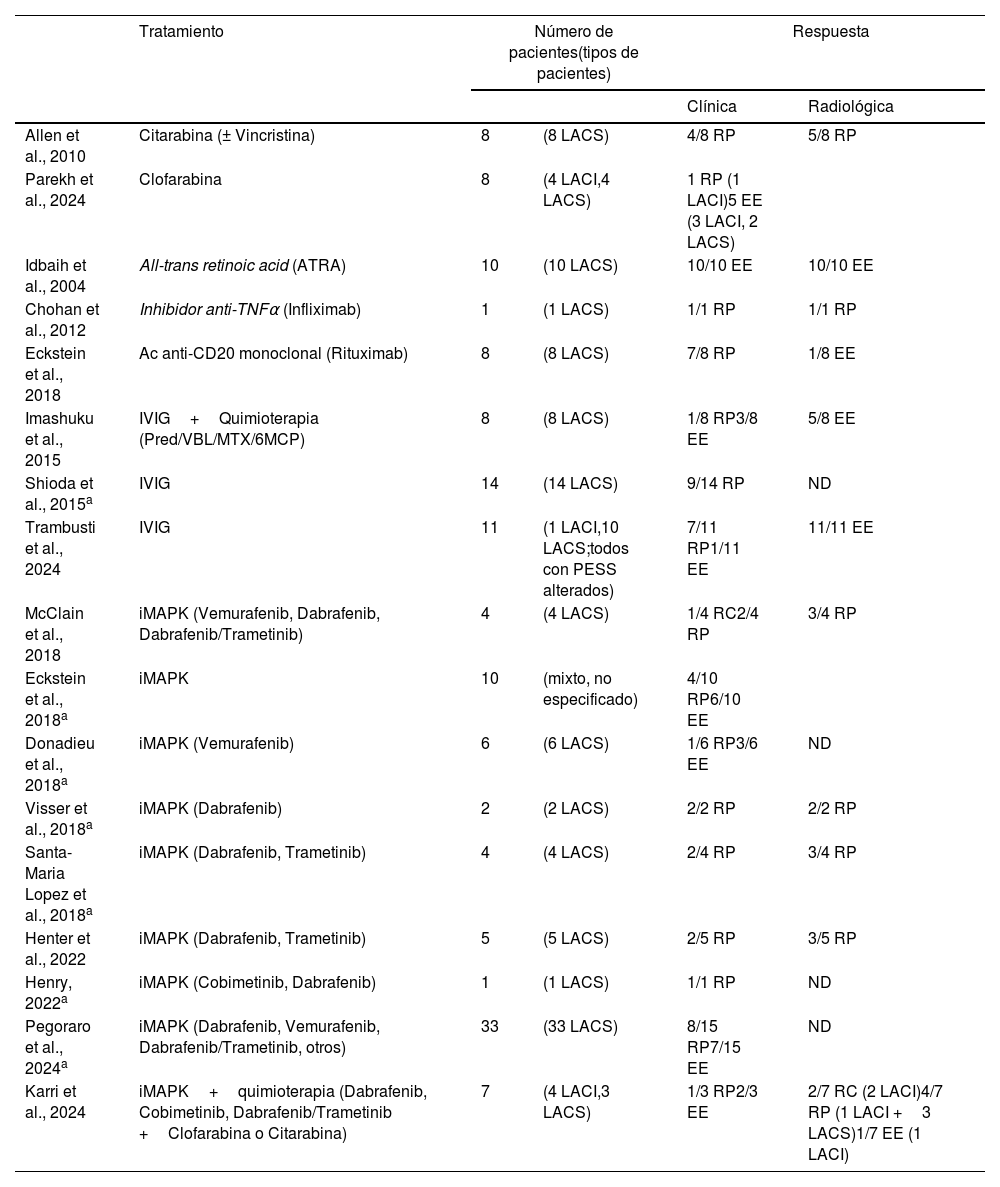
TratamientoEl manejo de la HCL-ND presenta desafíos significativos, y el tratamiento sigue siendo una cuestión compleja (tabla 1). Previo al cambio de modelo etiopatogénico de la enfermedad, las terapias convencionales mostraron, en la mayoría de los casos, estabilización de la enfermedad, lo que reflejaba el desconocimiento subyacente de la patología (ATRA, rituximab, infliximab). El uso de la quimioterapia (citarabina o clofarabina) ha mostrado mejoría clínico-radiológica en series pequeñas de pacientes37,38, y las inmunoglobulinas intravenosas (IVIG) producen respuestas objetivas, probablemente debido a su efecto inmunomodulador, y ambas siguen siendo utilizadas en la actualidad35,39.
Tratamientos utilizados para la HCL-ND
| Tratamiento | Número de pacientes(tipos de pacientes) | Respuesta | |||
|---|---|---|---|---|---|
| Clínica | Radiológica | ||||
| Allen et al., 2010 | Citarabina (± Vincristina) | 8 | (8 LACS) | 4/8 RP | 5/8 RP |
| Parekh et al., 2024 | Clofarabina | 8 | (4 LACI,4 LACS) | 1 RP (1 LACI)5 EE (3 LACI, 2 LACS) | |
| Idbaih et al., 2004 | All-trans retinoic acid (ATRA) | 10 | (10 LACS) | 10/10 EE | 10/10 EE |
| Chohan et al., 2012 | Inhibidor anti-TNFα (Infliximab) | 1 | (1 LACS) | 1/1 RP | 1/1 RP |
| Eckstein et al., 2018 | Ac anti-CD20 monoclonal (Rituximab) | 8 | (8 LACS) | 7/8 RP | 1/8 EE |
| Imashuku et al., 2015 | IVIG+Quimioterapia (Pred/VBL/MTX/6MCP) | 8 | (8 LACS) | 1/8 RP3/8 EE | 5/8 EE |
| Shioda et al., 2015a | IVIG | 14 | (14 LACS) | 9/14 RP | ND |
| Trambusti et al., 2024 | IVIG | 11 | (1 LACI,10 LACS;todos con PESS alterados) | 7/11 RP1/11 EE | 11/11 EE |
| McClain et al., 2018 | iMAPK (Vemurafenib, Dabrafenib, Dabrafenib/Trametinib) | 4 | (4 LACS) | 1/4 RC2/4 RP | 3/4 RP |
| Eckstein et al., 2018a | iMAPK | 10 | (mixto, no especificado) | 4/10 RP6/10 EE | |
| Donadieu et al., 2018a | iMAPK (Vemurafenib) | 6 | (6 LACS) | 1/6 RP3/6 EE | ND |
| Visser et al., 2018a | iMAPK (Dabrafenib) | 2 | (2 LACS) | 2/2 RP | 2/2 RP |
| Santa-Maria Lopez et al., 2018a | iMAPK (Dabrafenib, Trametinib) | 4 | (4 LACS) | 2/4 RP | 3/4 RP |
| Henter et al., 2022 | iMAPK (Dabrafenib, Trametinib) | 5 | (5 LACS) | 2/5 RP | 3/5 RP |
| Henry, 2022a | iMAPK (Cobimetinib, Dabrafenib) | 1 | (1 LACS) | 1/1 RP | ND |
| Pegoraro et al., 2024a | iMAPK (Dabrafenib, Vemurafenib, Dabrafenib/Trametinib, otros) | 33 | (33 LACS) | 8/15 RP7/15 EE | ND |
| Karri et al., 2024 | iMAPK+quimioterapia (Dabrafenib, Cobimetinib, Dabrafenib/Trametinib +Clofarabina o Citarabina) | 7 | (4 LACI,3 LACS) | 1/3 RP2/3 EE | 2/7 RC (2 LACI)4/7 RP (1 LACI +3 LACS)1/7 EE (1 LACI) |
Ac: anticuerpo; EE: enfermedad estable; iMAPK: inhibidor de la vía MAPK; IVIG: inmunoglobulinas intravenosas; LACI: LCH-associated abnormal CNS imaging; LACS: LCH-associated abnormal CNS symptoms; MTX: metotrexato; ND: no data; PE: progresión de enfermedad; PESS: potenciales evocados somatosensoriales; PR: respuesta parcial; Pred: prednisolona; RC: respuesta completa; 6MCP: 6-mercaptopurina; TNF: factor de necrosis tumoral; VBL: vinblastina.
Una cuestión clave en el tratamiento es determinar el momento adecuado para iniciar la terapia. En la era pre-molecular, la recomendación de un inicio precoz surgía de la ineficacia de los tratamientos para revertir la enfermedad; la mejoría ocurría generalmente en pacientes con corta evolución de la misma. En la actualidad, existen grupos que recomiendan iniciar el tratamiento en presencia de PESS alterados35, y la evaluación neurocognitiva comienza a tener un papel relevante como criterio clínico neuropsicológico. En pacientes con alteraciones únicamente por imagen (LACI) se opta por una actitud conservadora con seguimiento clínico-radiológico, dado que un porcentaje de estos pacientes puede que nunca empeoren ni muestren sintomatología. Sin embargo, ante progresión en la imagen, algunos grupos, incluso el protocolo LCH-IV de la Histiocyte Society (NCT02205762), sugieren valorar el inicio de tratamiento.
Tras el descubrimiento molecular se inició el uso de iMAPK también en la HCL-ND, mostrando respuestas clínico-radiológicas en hasta un 50-70% de los pacientes (tabla 1)15,36. La combinación de quimioterapia con iMAPK, recientemente reportada, muestra una tasa de respuesta similar40. A pesar del avance de los tratamientos dirigidos, los pacientes con una evolución prolongada, con probable pérdida neuronal y axonal más severa, siguen sin responder favorablemente. Esto refuerza la indicación de tratamiento precoz, para reducir o frenar la inflamación y prevenir el daño subsecuente. El tiempo de duración y los efectos tras la suspensión de estos tratamientos siguen siendo una incertidumbre.
En cuanto a guías de tratamiento, los grandes grupos colaborativos y grupos de expertos, en un esfuerzo de aunar directrices, sugieren recomendaciones de seguimiento y tratamiento (fig. 2). El consorcio norteamericano de HCL (NACHO) recomienda, en pacientes con LACI en progresión radiológica o en pacientes con sintomatología, el tratamiento con citarabina o iMAPK13,27. Cohen et al. realizan la misma recomendación, pero en casos con LACI recomiendan la evaluación neurocognitiva para considerar tratamiento e incluyen las IVIG dentro de las opciones terapéuticas1. Estudios pre-clínicos en modelos animales plantean nuevos potenciales tratamientos, como los inhibidores de CSFR119 o de Bcl-2 (agente senolítico)16, tanto en monoterapia como en combinación con iMAPK, que precisan de mayor investigación y ensayos clínicos.
ConclusionesLa forma neurodegenerativa de la HCL ha experimentado un cambio radical en su definición en los últimos años, y actualmente es considerada una forma de enfermedad activa originada por células presentes en el SNC que, por una activación constitutiva de la vía de señalización MAPK, producen neuroinflamación y posterior pérdida neuronal y axonal. Como factores de riesgo, la presencia de diabetes insípida central y la afectación ósea de órbita y/o base de cráneo siguen siendo considerados factores de riesgo de desarrollo de la forma ND, a los cuales se han unido la presencia de la mutación BRAFV600E. El seguimiento de los pacientes de riesgo y un diagnóstico temprano permitirían un inicio precoz del tratamiento. Las opciones de tratamiento aún son limitadas, pero los iMAPK han sido incorporados como armamento terapéutico a la quimioterapia y las inmunoglobulinas. Son necesarios estudios colaborativos a largo plazo para poder incorporar los nuevos descubrimientos a nivel diagnóstico y terapéutico en la población afecta de HCL-ND.
FinanciaciónEste trabajo no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los pacientes, familiares y profesionales que han participado en los estudios colaborativos de histiocitosis. A las asociaciones de familias afectas de HCL, por su apoyo incondicional y contagiosa motivación.
