



Presentamos el caso de un niño con retraso psicomotor y rasgos dismórficos afectado de una microdeleción 15q11.2 de 1.5 Mb de origen paterno diagnosticada mediante array-based comparative genomic hybridization. La deleción está comprendida entre los puntos de rotura BP1-BP2 de la región crítica descrita para los síndromes Prader-Willi/Angelman. Comparamos las características clínicas de nuestro paciente con las halladas en los 10 casos de deleción pura BP1-BP2 descritos hasta la fecha.
The case of a boy with psychomotor retardation and dysmorphic features is presented. He has a 1.5 Mb 15q11.2 microdeletion of paternal origin diagnosed by aCGH. The deletion is located between breakpoints BP1 and BP2 of the Prader-Willi/Angelman syndromes critical region. Clinical features in our patient fit well with those described in ten cases of pure BP1-BP2 deletion published to date.
La reciente introducción de nuevas técnicas diagnósticas como la aCGH (array-based comparative genomic hybridization) (citada en la práctica como «microarray cromosómico» o «cariotipo molecular») ha permitido, debido a su gran poder de resolución, un notable incremento en los diagnósticos de aquellas patologías que dependen de microdeleciones/microduplicaciones, así como la definición de un considerable número de nuevos síndromes y un mejor conocimiento de ciertos mecanismos etiopatogénicos en genética clínica. Debido a su potencial diagnóstico, algunos protocolos de consenso sugieren la sustitución de las técnicas habituales (cariotipo convencional y/o alta resolución, bandas subteloméricas, X-frágil) por aCGH como primera técnica genética diagnóstica en el estudio de pacientes neuropediátricos1.
Comunicamos el caso de un paciente de 4 años afectado de una microdeleción 15q11.2 de 1.5 Mb, indetectable con técnicas citogenéticas convencionales, comprendida entre los puntos de rotura BP1-BP2 de la región crítica para los síndromes Prader-Willi/Angelman (PWS/AS). Se ha sugerido que la haploinsuficiencia de los genes contenidos en la microdeleción sea la causante de ciertos rasgos fenotípicos (trastornos de conducta, alteraciones motoras y del habla) presentes en pacientes con PWS/AS con deleción tipo I (BP1-BP3) frente a pacientes con deleciones menores (tipo II) (BP2-BP3). Por otra parte, la misma microdeleción se ha detectado en pacientes con otros trastornos neuropsiquiátricos (retraso psicomotor/mental, epilepsia, esquizofrenia) como en nuestro caso, así como en población aparentemente normal.
Caso clínicoNiño de 4 años, hijo de padres sanos (de 35 y 37 años), no consanguíneos. Embarazo, parto y periodo neonatal sin incidencias. Tres hermanos de 11 años, 7 años y gemelo dicigoto sanos. Hermana paterna con artrogriposis congénita, epilepsia generalizada y trastorno de conducta desde los 9 años, desarrollando patología psiquiátrica (psicosis y trastorno de conversión) a los 28 años. Presenta malformación cerebral con displasia opercular bilateral y polimicrogiria silviana bilateral con extensión parieto-occipital.
Desde los primeros meses de vida refieren retraso psicomotor, con mayores dificultades en lenguaje (inicio con vocablos referenciales 1,5 años, frases simples 2,5 años) y a nivel motor (bipedestación con apoyo 12 meses, marcha libre a los 18 meses, dificultades para realizar pinza superior, manipulación tosca), asociando dificultades de atención. Bien adaptado a nivel social, con juego adecuado a su edad y buenas relaciones con otros niños.
A los 14 meses se realiza un estudio neuropsicológico, obteniéndose un coeficiente de desarrollo global de 0,75 (escala de desarrollo psicomotor de la primera infancia, Brunet Lézine revisada), iniciando estimulación y fisioterapia.
Durante el seguimiento, en un control realizado a los 3 años y 11 meses de edad, persisten problemas en la motricidad y el resto de áreas del desarrollo (escalas McCarthy de aptitudes y psicomotricidad). En lenguaje (test de aptitudes psicolingüísticas) obtiene resultados adecuados en todos los aspectos evaluados, con dificultades en integración visual y memoria secuencial visomotora. Se evalúa su capacidad cognitiva (test WPPSI, escala Wechsler de inteligencia para preescolar), comprobándose diferencias significativas en las escalas verbal (CI V: 100) y manipulativa (CI M: 80), con un CI total de 88.
En la escuela refieren problemas en la motricidad fina (dificultades en trazo y para escribir números y letras) y gruesa (corre, sube y baja escaleras con dificultad), buenas relaciones a nivel social, autónomo, iniciando juego más elaborado, y persistiendo dificultades de atención.
Respecto a los problemas de atención, se ha evaluado a través de los padres la presencia de conductas de inatención e hiperactividad (escalas de Conners), comprobándose la presencia de signos y síntomas suficientes para diagnosticarle de un trastorno por déficit de atención e hiperactividad (TDAH).
Exploración física: perímetro cefálico en percentil 10, con talla y peso normal, angioma en el tercio inferior de la espalda de 2-3mm, facies peculiar alargada con frente ancha, discreto hipertelorismo, desviación ocular antimongoloide, pabellones auriculares grandes y de implantación baja, raíz nasal ancha, filtrum largo, labio superior fino y evertido, micrognatia, discreta hipotonía axial y de cintura escapular, con resto de la exploración neurológica normal. Analítica sanguínea, hormonas tiroideas, potenciales evocados auditivos de tronco (PEAT) y resonancia magnética cerebral sin hallazgos.
Estudios genéticos: un estudio citogenético convencional en sangre periférica no detectó (nivel de bandeo GTG 550) anomalías numéricas y/o estructurales. El estudio X-Frágil fue negativo. Se realizó un microarray CGH Cytoarray (44K) (Agilent Technologies, Santa Clara, CA, Esatados Unidos) que detectó una microdeleción en la citobanda 15q11.2 de 1,5 Mb (chr 15: 19108924-20636678) (fig. 1). Esta deleción, situada por debajo del poder de resolución de la citogenética convencional, afecta a cuatro genes: NIPA1, NIPA2, CYFIP1, TUBGCP5. La microdeleción se encuentra comprendida entre los puntos de rotura BP1-BP2 de la región crítica descrita para PWS/AS (fig. 2). El estudio familiar demuestra que la deleción es de origen paterno, siendo también dos hermanos del paciente portadores, todos ellos asintomáticos. La hermana paterna, con patología neuropsiquiátrica, no es portadora de la microdeleción.
Discusión que muestra la deleción del cromosoma 15.")
.")
La región de los brazos largos del cromosoma 15 comprendida entre las citobandas q11 y q13 contiene secuencias duplicadas, al igual que otras zonas del genoma2,3. La presencia de estas secuencias low-copy repeat en puntos de rotura recurrentes (BP1, BP2, BP3) confiere una gran inestabilidad, siendo frecuentes las deleciones, duplicaciones y la formación de pequeños cromosomas marcadores, ya que estas secuencias repetidas pueden dar lugar a una incorrecta recombinación meiótica por recombinación homóloga no alélica que puede ser intercromosómica (entre cromosomas homólogos) o intracromosómica (en el mismo cromosoma)3.
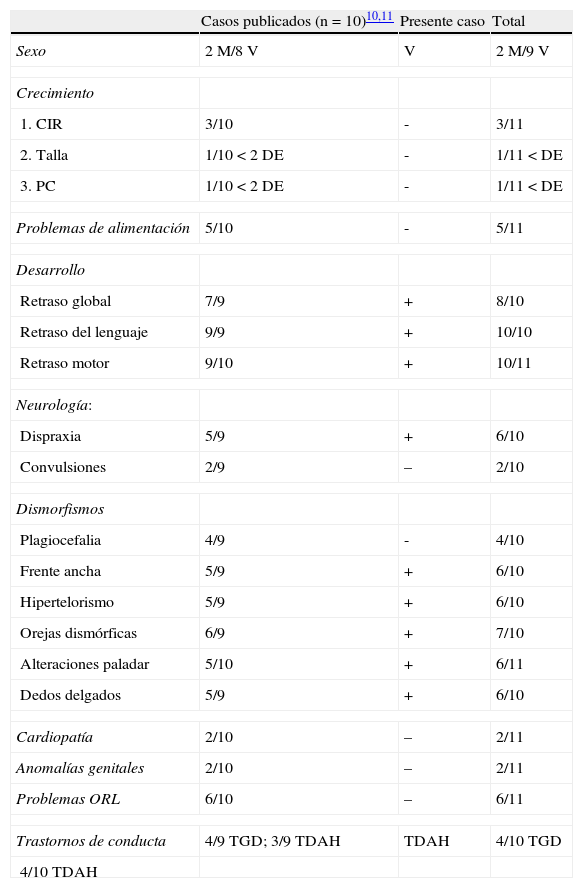
Las deleciones entre BP1-BP3 y BP2-BP3 dan lugar a PWS/AS, dependiendo de que la deleción ocurra en el cromosoma 15 paterno (PWS) o materno (AS), siendo éste el mecanismo más frecuente (aproximadamente el 70% de los casos) para ambos síndromes. Algunos estudios4–7 señalan diferencias fenotípicas entre los dos subtipos de PWS por deleción: aquellos pacientes con PWS con deleción tipo I (BP1-BP3) presentan mayores trastornos conductuales (conductas obsesivo-compulsivas, peor conducta adaptativa) y menor habilidad intelectual (peor integración visual-motora, problemas del lenguaje) que los pacientes con PWS con deleción tipo II (BP2-BP3). Estas diferencias fenotípicas también existen al comparar pacientes con PWS con deleción tipo I con pacientes con PWS por disomía uniparental materna. Similares diferencias se han descrito en pacientes AS con los dos tipos de deleción y con disomía uniparental paterna8,9. En la región comprendida entre BP1 y BP2 se han identificado 4 genes (NIPA1, NIPA2, CYFIP1, TUBGCP5), que no están sujetos a imprinting (expresión diferencial de un gen dependiendo de su origen materno o paterno) y, por tanto, con una expresión bialélica. TUBGCP5 se expresa fundamentalmente a nivel de los núcleos subtalámicos cerebrales, relacionados con TDAH y conducta obsesivo-compulsiva. NIPA1 y NIPA2 tienen amplia expresión en el sistema nervioso y codifican proteínas que actúan como transportadores de membrana. CYFIP1 codifica una proteína que interactúa con FMRP (fragile X mental retardation protein) e interviene en el desarrollo y mantenimiento de estructuras neuronales10. La descripción reciente de 10 pacientes con deleción BP1-BP2 pura10,11 (tabla 1), semejante a la descrita en nuestro caso, sin un fenotipo PWS/AS pero con retraso motor y en el área del lenguaje, dismorfias y trastornos de conducta, sugiere que la haploinsuficiencia de los cuatro genes citados sería la causante del cuadro clínico de este posible nuevo síndrome de microdeleción 15q11.2.
Manifestaciones clínicas de la microdeleción 15q11.2
| Casos publicados (n = 10)10,11 | Presente caso | Total | |
| Sexo | 2 M/8 V | V | 2 M/9 V |
| Crecimiento | |||
| 1. CIR | 3/10 | - | 3/11 |
| 2. Talla | 1/10<2 DE | - | 1/11<DE |
| 3. PC | 1/10<2 DE | - | 1/11<DE |
| Problemas de alimentación | 5/10 | - | 5/11 |
| Desarrollo | |||
| Retraso global | 7/9 | + | 8/10 |
| Retraso del lenguaje | 9/9 | + | 10/10 |
| Retraso motor | 9/10 | + | 10/11 |
| Neurología: | |||
| Dispraxia | 5/9 | + | 6/10 |
| Convulsiones | 2/9 | – | 2/10 |
| Dismorfismos | |||
| Plagiocefalia | 4/9 | - | 4/10 |
| Frente ancha | 5/9 | + | 6/10 |
| Hipertelorismo | 5/9 | + | 6/10 |
| Orejas dismórficas | 6/9 | + | 7/10 |
| Alteraciones paladar | 5/10 | + | 6/11 |
| Dedos delgados | 5/9 | + | 6/10 |
| Cardiopatía | 2/10 | – | 2/11 |
| Anomalías genitales | 2/10 | – | 2/11 |
| Problemas ORL | 6/10 | – | 6/11 |
| Trastornos de conducta | 4/9 TGD; 3/9 TDAH | TDAH | 4/10 TGD |
| 4/10 TDAH | |||
TDAH: trastorno por déficit de atención e hiperactividad; TGD: trastorno generalizado del desarrollo.
Estudios recientes relacionan la microdeleción 15q11.2 con la epilepsia idiopática generalizada12,13 o con la esquizofrenia14, sugiriendo que la deleción BP1-BP2 en la región 15q11.2 confiere susceptibilidad genética a una amplia gama de trastornos neuropsiquiátricos, aunque con una penetrancia incompleta (tabla 2).
De los 11 pacientes descritos10,11 con deleción pura BP1-BP2 (incluido el presente caso), 6 son de origen paterno, 3 materno y 2 de novo (el cromosoma delecionado es el materno). Analizando los 9 casos heredados, 2 de los padres portadores están afectados y en 4 de los 9 hay antecedentes familiares de distintos trastornos neuropsiquiátricos compatibles con los descritos en la microdeleción BP1-BP2.
El caso familiar que describimos confirma la variable expresividad observada en otros casos similares, planteando ciertos problemas en cuanto al consejo genético y a la valoración clínica de sujetos asintomáticos (como los hermanos de nuestro paciente), así como un posible diagnóstico prenatal. Por otra parte, el cuadro clínico que presenta nuestro paciente es superponible al de los otros casos descritos, lo que indica la existencia de un nuevo síndrome de microdeleción 15q11.2.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
