


La insuficiencia suprarrenal primaria (ISP) es una enfermedad rara en niños, caracterizada por la incapacidad en la producción de glucocorticoides y/o mineralocorticoides. Las manifestaciones clínicas son inespecíficas e insidiosas. Debemos conocer esta enfermedad para poder realizar un diagnóstico precoz, ya que el correcto manejo de la enfermedad puede salvar vidas.
DiseñoSe ha realizado un estudio multicéntrico y retrospectivo, registrándose todos los pacientes diagnosticados de ISP (menores de 18 años) en los últimos 30 años de cinco hospitales españoles.
ObjetivosDeterminar la etiología, signos, síntomas y alteraciones analíticas en la insuficiencia suprarrenal primaria en la edad pediátrica.
ResultadosVeintinueve pacientes fueron diagnosticados de ISP, con una mediana de 5,6 años. Se logró emitir un diagnóstico etiológico en 23 pacientes (79,3%): encontramos 8 casos de adrenoleucodistrofia ligada al cromosoma X (27,6%), 6 adrenalitis autoinmunes (20,7%), 4 hipoplasias suprarrenales congénitas ligadas al X (13,8%), 2 síndromes de resistencia a la ACTH (6,9%), 2 pacientes con síndrome de Pearson (6,9%) y un paciente con síndrome de Allgrove (3,4%). En los otros seis pacientes, la etiología es desconocida por el momento. Dieciséis pacientes (55,2%) debutaron en forma de crisis adrenal. Veinte pacientes (69%) precisaron tratamiento combinado (hidrocortisona y fludrocortisona).
ConclusionesLa astenia, la hiperpigmentación cutánea y la hiponatremia fueron el síntoma, el signo y la alteración electrolítica más frecuentes al debut, aunque su ausencia no descarta una ISP. La ACTH (hormona adrenocorticotropa) permanece elevada a pesar de un correcto tratamiento con glucocorticoides.
Primary adrenal insufficiency (PAI) in children is a rare condition characterized by deficient production of glucocorticoids and/or mineralocorticoids. The clinical manifestations are nonspecific and insidious. Providers need to know about this disorder to be able to make an early diagnosis, as appropriate management can be life-saving.
MethodsWe conducted a multicentre retrospective study including every patient aged less than 18 years given a diagnosis of PAI in the last 30 years at 5 Spanish hospitals.
ObjectivesThe objective was to determine the aetiologies, signs, symptoms and laboratory findings of PAI in the paediatric age group.
ResultsTwenty nine patients received a diagnosis of PAI at a median age of 5.6 years. An aetiological diagnosis was established in 23 patients (79.3%): X-linked adrenoleukodystrophy in 8 (27.6%), autoimmune adrenalitis in 6 (20.7%), X-linked adrenal hypoplasia congenita in 4 (13.8%), adrenocorticotropic hormone (ACTH) resistance syndrome in 2 (6.9%), Pearson syndrome in 2 (6.9%) and Allgrove syndrome in 1 (3.4%). In the remaining 6 patients, no clear aetiology was identified. Sixteen patients (55.2%) had onset with an adrenal crisis. Twenty patients (69%) needed combination therapy (hydrocortisone and fludrocortisone).
ConclusionsAsthenia, hyperpigmentation and hyponatraemia were the most prevalent sign, symptom and electrolyte abnormality at onset of PAI, although their absence does not rule out this disease. The elevation of ACTH persists despite adequate glucocorticoid replacement therapy.

La insuficiencia suprarrenal primaria (ISP) consiste en la incapacidad de las propias glándulas suprarrenales de producir hormonas adrenocorticales (glucorticoides y mineralocorticoides).
Se trata de una entidad poco frecuente, situándose la prevalencia en 82 a 144 casos por millón de habitantes y la incidencia en 4 a 6 casos por millón de habitantes, con un predominio hacia el sexo femenino1-3, siendo desconocida su incidencia en la infancia.
La causa más frecuente de ISP de presentación neonatal es la hiperplasia suprarrenal congénita (HSC), debida al déficit de 21-hidroxilasa. Son escasas las casuísticas en la infancia, como la que presentaremos a continuación, que valoran etiologías minoritarias como las formas autoinmunes, la resistencia corticotropa u otros defectos genéticos. Hasta la fecha, en España, sólo hemos encontrado otra más4.
La adrenalitis autoinmune es la causa más frecuente de ISP en adultos, siendo responsable de un 80% de los casos, existiendo un claro predominio hacia el sexo femenino5. Es mucho menos frecuente en niños, justificando tan solo un 10-15% de los casos5.
Se puede presentar de forma aislada o asociada a otras enfermedades como hipoparatiroidismo, candidiasis mucocutánea u otras enfermedades autoinmunes, formando parte de un síndrome pluriglandular autoinmune (SPA).
La adrenoleucodistrofia ligada al X (ALD-X) es un trastorno peroxisomal, cuya incidencia es de 1 por cada 14.700 recién nacidos6. Se debe a mutaciones en el gen ABCD1 (Xq28) que codifica una proteína transmembrana (ALDP) implicada en el transporte de los ácidos grasos de cadena muy larga (AGCML) desde el citosol hasta el peroxisoma, donde se degradan. El acúmulo de los AGCML produce desestabilización de la vaina de mielina, responsable de la sintomatología neurológica, daños a nivel de la corteza suprarrenal e hipogonadismo, al acumularse en las células de Leydig.
La hipoplasia suprarrenal congénita se debe a alteraciones del gen NR0B1, que codifica la proteína DAX1, situada en la región Xp21.2. La herencia suele ser recesiva ligada al cromosoma X. Es un trastorno poco frecuente, que ocurre en una frecuencia menor a 1 de cada 12.500 recién nacidos7. Cursa con insuficiencia suprarrenal primaria, pubertad retrasada y alteración en la espermatogénesis en la etapa adulta.
Debe sospecharse en varones, tras descartarse la HSC y la ALD-X, sobre todo si existen antecedentes familiares de infertilidad en varones.
El síndrome de resistencia a la ACTH o deficiencia de glucocorticoides familiar (FGD) es una enfermedad infrecuente, de prevalencia desconocida y herencia autosómica recesiva. Cursa con un déficit aislado de glucocorticoides por falta de respuesta de las células de la zona fasciculada de la corteza suprarrenal a la ACTH. Se debe a defectos en el receptor de la ACTH (MC2R) o en su proteína accesoria (MRAP) que transmite la señal de la ACTH al interior de las células8. Debemos sospecharla ante pacientes con hipoglucemias de repetición e hiperpigmentación cutáneo-mucosa. Apoyan el diagnóstico los antecedentes familiares de consanguinidad o muertes prematuras sin filiar.
El síndrome de Pearson es una enfermedad rara, secundaria a una deleción en el ADN mitocondrial. Su incidencia es de 1 por millón de recién nacidos9. Se caracteriza por la existencia de anemia sideroblástica asociada a neutropenia y trombopenia, disfunción pancreática exocrina y tubulopatía renal. Puede asociar diabetes, insuficiencia suprarrenal primaria, hipoparatiroidismo y retraso del crecimiento.
El síndrome de Allgrove (AS), también conocido como síndrome de triple A, se caracteriza por presentar alacrimia, acalasia e insuficiencia adrenal secundaria a resistencia a la ACTH. Asocia neuropatía periférica motora y sensitiva, disautonomía, atrofia óptica y ataxia. La prevalencia es desconocida. Se hereda de forma autosómica recesiva por mutaciones en el gen AAAS (12q13.13) que codifica la proteína ALADIN10.
A menudo la clínica derivada del déficit combinado de cortisol y mineralocorticoides suele instaurarse de manera progresiva, siendo los síntomas inespecíficos lo que dificulta el diagnóstico.
El déficit de glucocorticoides se manifiesta con astenia, anorexia y tendencia a la hipoglucemia, debidos a los efectos de la falta de cortisol sobre el metabolismo energético (disminución de la gluconeogénesis y la glucogenólisis), además puede producir hipotensión ortostática, por disminución de la sensibilidad a las catecolaminas, y molestias gastrointestinales debidos al aumento de ácido clorhídrico y pepsina en la luz intestinal. Es habitual la pérdida de peso.
El descenso del cortisol condiciona un aumento de CRH (hormona liberadora de corticotropina), ACTH y otros péptidos derivados de la proopiomelanocortina, como la β-lipotropina, que tiene actividad estimulante de los melanocitos, causando hiperpigmentación de piel y mucosas, característica de la enfermedad de Adisson.
El déficit de mineralocorticoides es responsable de la pérdida salina por hipoaldosderonismo, que causará hipovolemia, hiponatremia con natriuresis aumentada, hiperpotasemia y acidosis metabólica. Es típica la avidez por la sal. En la analítica encontraremos niveles bajos de aldosterona con elevación de renina de forma compensatoria.
Es de vital importancia su sospecha, ya que cualquier tipo de estrés (infecciones, traumatismos, cirugías…) puede provocar un cuadro de insuficiencia suprarrenal aguda, conocido como crisis adrenal, cuyas manifestaciones clínicas corresponden a un shock hipovolémico con hiponatremia, hiperpotasemia, acidosis e hipoglucemia.
La incidencia estimada en Europa de crisis adrenales es de 6 a 8 pacientes por cada 100 habitantes/año, ocurriendo un 70% de ellas en los primeros 10 años de vida11,12. La tasa de mortalidad es de 0,5 casos por cada 100 pacientes/año2. Se trata, por tanto, de un cuadro grave que precisa atención médica urgente.
Consideramos de alto interés el estudio, dada la escasa prevalencia en pediatría, el alto grado de sospecha requerido para su diagnóstico y la alta mortalidad de la crisis suprarrenal.
ObjetivosEl objetivo general fue conocer las causas de insuficiencia suprarrenal primaria en pediatría. Los objetivos específicos fueron: conocer datos epidemiológicos de las distintas etiologías, recopilar los síntomas, signos y hallazgos analíticos presentes al debut, conocer la frecuencia del debut en forma de crisis suprarrenal, determinar el valor de la ACTH en la monitorización del tratamiento y recopilar cuántos pacientes precisaron tratamiento combinado (glucocorticoides y mineralocorticoides).
Pacientes y métodosPacientes. Se realizó un estudio descriptivo de una serie de casos, multicéntrico, en el que se incluyeron a todos los pacientes diagnosticados de ISP en la infancia, durante los últimos 30 años (desde enero de 1990 hasta diciembre de 2020), en cinco hospitales españoles.
Se incluyeron a todos los pacientes con sospecha de ISP y confirmación posterior, cuyo debut se comprende desde el nacimiento hasta los 18 años. Su historia clínica debía ser compatible y presentar niveles séricos de ACTH elevados y/o niveles de cortisol disminuidos.
Se excluyeron a todos los pacientes fallecidos con imposibilidad de recoger los datos, los casos de hiperplasia suprarrenal congénita y aquellos cuyo origen fue iatrogénico, tumoral, infeccioso y/o traumático.
Métodos. Los datos se recopilaron a partir de historias clínicas (formato papel y/o digital).
El análisis de datos se realizó con el programa SPSS 25.0 (IBM, NY, EE. UU.). Los resultados se expresan con frecuencias y porcentajes (variables cualitativas) y con mediana, valor mínimo y máximo (variables cuantitativas). Las comparaciones de las proporciones se realizan con la prueba de Chi cuadrado o exacta de Fisher. Las comparaciones de grupos en variables cuantitativas u ordinales se realizan con las pruebas de Kruskal-Wallis y U de Mann-Whitney. Se consideran valores significativos los valores de p<0,05.
Aspectos éticos. La propuesta del trabajo fue aprobada por el comité ético de investigación clínica local.
ResultadosCausas de insuficiencia suprarrenal y epidemiologíaLa muestra final consta de 29 pacientes, 22 (75%) varones y 7 (25%) mujeres. Se logró emitir un diagnóstico etiológico en 23 (79,3%): encontramos ocho casos de adrenoleucodistrofia ligada al X (27,6%), seis adrenalitis autoinmunes (20,7%), cuatro hipoplasias suprarrenales congénitas secundarias a alteraciones en la proteína DAX1 (13,8%), dos síndromes de resistencia a la ACTH (6,9%), dos pacientes con síndrome de Pearson (6,9%) y un paciente con síndrome de Allgrove (3,4%).
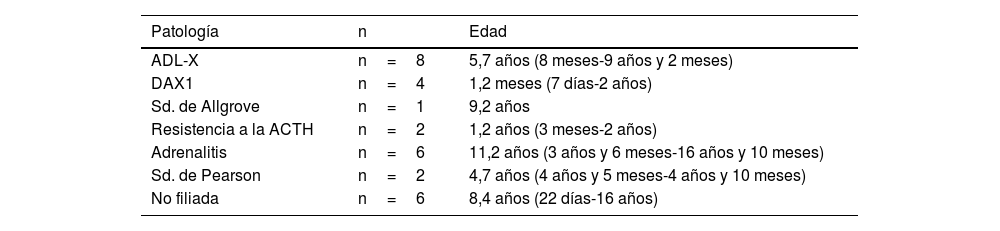
La edad en el momento del diagnóstico está comprendida entre los siete días de vida y los 16 años, siendo la mediana de 5,6 años. La tabla 1 recoge la edad al debut de los pacientes en función de la etiología.
Edad al debut de los pacientes según la etiología de la ISP
| Patología | n | Edad |
|---|---|---|
| ADL-X | n=8 | 5,7 años (8 meses-9 años y 2 meses) |
| DAX1 | n=4 | 1,2 meses (7 días-2 años) |
| Sd. de Allgrove | n=1 | 9,2 años |
| Resistencia a la ACTH | n=2 | 1,2 años (3 meses-2 años) |
| Adrenalitis | n=6 | 11,2 años (3 años y 6 meses-16 años y 10 meses) |
| Sd. de Pearson | n=2 | 4,7 años (4 años y 5 meses-4 años y 10 meses) |
| No filiada | n=6 | 8,4 años (22 días-16 años) |
ACTH: hormona adrenocorticotrópica; ADL-X: adrenoleucodisdrofia ligada al X; DAX1: hipoplasia suprarrenal congénita; ISP: insuficiencia suprarrenal primaria; Sd: síndrome.
Tres mujeres y tres varones fueron diagnosticados, detectándose en todos los casos la presencia de anticuerpos anti corteza suprarrenal.
Cuatro de ellos (67%) presentaron una crisis adrenal en el debut, mientras que la clínica de los otros dos fue más larvada, siendo los motivos de consulta candidiasis mucocutánea-retraso puberal, y pérdida de peso-decaimiento.
En el 66,7% de ellos, se halló otro tipo de autoinmunidad: anticuerpos anti tiroideos, anti hipófisis y anti ovario.
En la mitad de los pacientes la insuficiencia suprarrenal es aislada por el momento, los otros tres pertenecen a un síndrome pluriglandular autoinmune (SPA); dos de ellos asocian un hipotiroidismo primario.
Adrenoleucodistrofia ligada al X (ADL-X)Encontramos ocho varones, dos de los pacientes eran hermanos, y otros dos, tenían el antecedente de un hermano fallecido por esta patología.
En esta serie, el 87,5% de los casos se diagnosticó a raíz de una clínica neurológica larvada, destacando la alteración en el comportamiento en cinco pacientes (62,5%), seguida de las alteraciones de la marcha y del lenguaje en tres de ellos (37,5%). Un 25% de los pacientes presentaban déficit visual, auditivo o hipotonía. Un paciente se diagnosticó a raíz de un cuadro de deshidratación hiponatrémica (crisis adrenal).
La concentración plasmática de los AGCML (ácidos grasos de cadena muy larga) estaba elevada en todos los casos.
El estudio genético se llevó a cabo en el 50% de los pacientes, confirmando la alteración en el gen ABCD1. Las variantes patogénicas encontradas fueron: c.521A>G (p.Tyr174Cys) y c.901-3C>G.
Durante el seguimiento, se produjo el éxitus de dos pacientes.
Hipoplasia suprarrenal congénitaSe trata de cuatro varones. Como se refleja en la tabla 1, fueron los pacientes que debutaron a una edad más temprana, dos de ellos incluso, durante el periodo neonatal.
En todos se confirmó, mediante técnicas de diagnóstico molecular, las alteraciones en el gen NR0B1, que codifica la proteína DAX-1. Las variantes patogénicas encontradas fueron las siguientes: c.707G>A (p.Trp236X), c.773C>A (p.Ala258Asp) y c.315G>A (p.Trp105X).
Dos de los pacientes fueron diagnosticados de hipogonadismo hipogonadotropo durante su seguimiento y recibieron tratamiento con testosterona intramuscular.
Síndrome de resistencia a la ACTH o deficiencia de glucocorticoides familiar (FGD)Dos mujeres fueron diagnosticadas. En una de las pacientes se confirmó la etiología al encontrar en el estudio genético dos mutaciones en heterocigosis en el gen del receptor de la ACTH (MC2R). Las variantes encontradas fueron: MC2R:c.320A>G (p.Asp107Gly) y MC2R:c.433C>T (p.Arg145Cys).
La otra paciente carece de estudio genético, pero se sospecha esta entidad ante la presencia de consanguinidad entre los progenitores y la ausencia de déficit mineralocorticoideo.
Llama la atención que ambas pacientes debutaron con crisis convulsivas secundarias a hipoglucemia grave.
Síndrome de PearsonEncontramos dos varones que, como parte de su enfermedad de base, desarrollaron pancitopenia, insuficiencia pancreática exocrina, tubulopatía, retraso de crecimiento postnatal y retraso psicomotor. Uno desarrolló un hipotiroidismo primario no autoinmune, y llama la atención que ambos pacientes fueron diagnosticados de un déficit de hormona de crecimiento.
Síndrome de AllgroveEl único paciente es un varón que debutó con una convulsión secundaria a una hipoglucemia grave. Previo a la ISP había sido diagnosticado de acalasia, alacrimia y alteraciones neurológicas: PEV (potenciales evocados visuales) y PEATC (potenciales evocados auditivos del tronco cerebral) alterados y atrofia muscular espinal distal. Ante la presencia de la tríada característica, se emitió el síndrome de Allgrove, aunque no se confirmó genéticamente.
Síntomas, signos y hallazgos analíticos presentes al debutLa astenia fue el síntoma predominante, referido por 16 pacientes (55%), seguido de los vómitos y la pérdida de peso que mostraron 14 pacientes (48,3%), y la anorexia, referida por 11 (37,9%).
Cuatro pacientes presentaron crisis convulsivas al inicio, tres secundarias a hipoglucemia, con niveles de glucemia en sangre de 30, 14 y 8mg/dL, respectivamente, y uno secundario a una hiponatremia grave, cuya cifra de sodio en sangre era de 104 mEq/L.
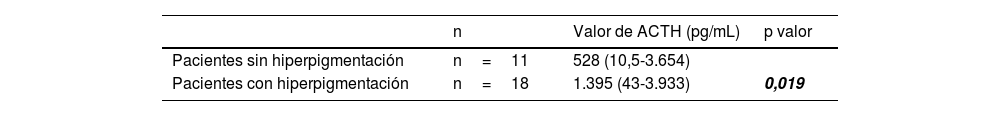
La hiperpigmentación fue el signo clínico más repetido, estando presente en 18 pacientes (62,1%). Los valores de ACTH al diagnóstico fueron más elevados en los pacientes que presentaban hiperpigmentación (tabla 2).
La hiponatremia fue la alteración electrolítica más frecuente, presente en 19 pacientes (65,5%). La mediana de natremia fue de 129,5 mEq/L (103-141).
La hiperpotasemia sólo se objetivó en siete pacientes (24,1%). La mediana de potasio en sangre fue de 5 mEq/L (3,8-7,8).
En cuanto a los niveles de glucosa en sangre, 10 pacientes (34,5%) presentaron niveles inferiores a 60mg/dL, seis de ellos, mostraban cifras de menores de 50mg/dL. La mediana de glucemia fue de 71,5mg/dL (8-111).
El nivel de ACTH en sangre se encontró elevado al debut en la gran mayoría de los casos (93,1%). La mediana de los valores de la ACTH fue de 1.100 pg/mL (10,5-3.933). No se encontraron diferencias entre los niveles de ACTH y las diferentes etiologías.
Se detectaron niveles de cortisol en sangre inferiores a 5 mcg/dL en 17 pacientes (58,6%), entre 5-9 mcg/dL en 7 (24,1%) y mayores de 9 mcg/dL en 5 casos (17,2%). En todos los pacientes con niveles de cortisol superiores a 5 mcg/dL, la ACTH estaba elevada.
No se ha podido demostrar la existencia de una correlación negativa entre los niveles de cortisol y los niveles de ACTH al diagnóstico.
Los niveles de aldosterona se encontraron disminuidos en 17 pacientes (58,6%); sin embargo, la ARP (actividad de renina plasmática) sólo se encontró elevada en 11 casos (37,9%).
Debut en forma de crisis suprarrenalDesde el punto de vista clínico el debut fue agudo (crisis suprarrenal) en 16 casos (55,2%), aunque sólo se detectó hipotensión arterial en 11 de ellos.
La tabla 3 muestra los pacientes que debutaron con crisis suprarrenal en función de la etiología.
Porcentaje de pacientes con debut en forma de crisis adrenal en función de la etiología de la ISP
| ADL-X | DAX1 | Sd. de Allgrove | Resistencia a la ACTH | Adrenalitis autoinmune | Sd. de Pearson | No filiada |
|---|---|---|---|---|---|---|
| n=8 | n=4 | n=1 | n=2 | n=6 | n=2 | n=6 |
| 1 (12,5%) | 4 (100%) | 1 (100%) | 2 (100%) | 4 (66,7%) | 0% | 4 (66,7%) |
ACTH: hormona adrenocorticotrópica; ADL-X: adrenoleucodisdrofia ligada al X; DAX1: hipoplasia suprarrenal congénita; ISP: insuficiencia suprarrenal primaria; Sd: síndrome.
No se observaron diferencias significativas entre la existencia de crisis suprarrenales y la edad al diagnóstico.
De los pacientes que desarrollaron una crisis adrenal al debut, seis (37,5%) presentaron una de las alteraciones analíticas de la tríada asociada a la crisis adrenal (hiponatremia, hiperpotasemia e hipoglucemia), siete (43,8%) presentaron dos alteraciones, y tan solo tres (18,7%) asociaban todas ellas.
Monitorización durante el seguimiento de los niveles de ACTHLos niveles de ACTH mostraron fluctuaciones durante el seguimiento, llegándose a normalizar o incluso suprimir transitoriamente en el 69% de los casos. La última determinación se encontraba elevada en el 66% de los pacientes. No se encontró diferencias entre este hallazgo y las diferentes etiologías.
TratamientoVeinte pacientes (69%) recibieron tratamiento combinado con hidrocortisona y nueve αfludrocortisona, mientras que nueve (31%) fueron tratados con glucocorticoides en monoterapia (las dos pacientes con síndrome de resistencia a la ACTH, los dos pacientes con síndrome de Pearson y cinco de los pacientes con adrenoleucodistrofia).
DiscusiónLas causas de ISP más frecuentes de este estudio son: la adrenoleucodistrofia ligada al X, la adrenalitis autoinmune y la hipoplasia suprarrenal congénita. En nuestra serie predomina la adrenoleucodistrofia ligada al X, coincidiendo con otro artículo publicado recientemente13; sin embargo, otros trabajos siguen registrando el origen autoinmune como etiología mayoritaria14-16.
Destaca, como etiología con debut más temprano, la hipoplasia suprarrenal congénita (mediana de edad al diagnóstico de 1,2 meses), mientras que la adrenalitis autoinmune afecta a los pacientes de mayor edad (mediana de edad al diagnóstico de 11,2 años), coincidiendo con la literatura7,15.
La ratio varón/mujer es de 1/0,3, coincidiendo el predominio masculino con otras series de casos publicadas con pacientes pediátricos4,13,14.
Según nuestro trabajo, la astenia, la hiperpigmentación y la hiponatremia fueron el síntoma, el signo y la alteración electrolítica más frecuentes, aunque su ausencia, no descarta una ISP.
La hiperpigmentación cutánea es el signo clínico guía que nos orienta al diagnóstico, pero no siempre está presente. Estudios publicados reflejan la presencia de hiperpigmentación en el 90% de los pacientes17-19 no siendo tan frecuente en nuestra muestra (62,1%). Puede ser muy sutil al inicio del cuadro o en las primeras seis semanas de vida17, por lo que esta podría ser la explicación a la ausencia de hiperpigmentación en tres de nuestros pacientes, cuyo debut fue a los 7, 22 y 24 días de vida.
La hiponatremia estuvo presente en el 65,5% de los pacientes, aunque este hallazgo puede ser aún mayor (90%)20,21.
La hiperpotasemia es escasa en nuestra muestra (24,1%) en comparación con otras series publicadas, presente hasta en el 50% de los casos16,20. Su ausencia puede justificarse en caso de vómitos abundantes, dado que la pérdida de ácido clorhídrico, conlleva una alcalosis metabólica y secundariamente, una hipopotasemia, aunque en nuestra muestra, no encontramos relación entre los niveles de potasio y la presencia de vómitos.
Un 55,2% de nuestros pacientes debutaron con una crisis suprarrenal aguda. Se define crisis adrenal en pediatría como un deterioro agudo del estado de salud que curse con alteración hemodinámica (hipotensión arterial, relleno capilar enlentecido o taquicardia inapropiada para la edad) o una alteración electrolítica grave (hiponatremia o hiperpotasemia) o hipoglucemia que no se atribuya a ninguna otra causa. El diagnóstico se confirma cuando las alteraciones se revierten tras la administración de glucocorticoides22.
Como refleja nuestra serie, no es necesario presentar hipotensión arterial para el diagnóstico de una crisis suprarrenal, ya que más de la mitad de nuestros pacientes debutaron en forma de crisis adrenal, pero sólo se objetivó hipotensión arterial en el 37,9%. Igualmente, no debemos esperar a la afectación conjunta de los valores de sodio, potasio y glucemia para sospechar una crisis adrenal.
La ACTH no es un buen parámetro para la monitorización del tratamiento. Los valores de la ACTH deben permanecer elevados, como refleja nuestra muestra, si los niveles están dentro de la normalidad, es posible que el paciente esté sobretratado3,17. Igualmente, no debemos modificar el tratamiento guiándonos por el grado de hiperpigmentación17. Para el ajuste del corticoide es preciso evaluar la ganancia de peso, el crecimiento y la clínica.
En nuestra serie, el 69% precisaron tratamiento combinado (glucocorticoide y mineralocorticoide). El resto de los pacientes, no mostraban afectación del eje mineralocorticoideo, esto se debe a que la zona glomerular de la corteza suprarrenal suele afectarse en menor medida que la fascicular y la reticular6.
En conclusión, debemos mantenernos alerta para reconocer los síntomas y signos de una insuficiencia suprarrenal primaria, ya que lograremos un diagnóstico precoz, previniendo el desarrollo de crisis adrenales. Además, si conocemos las diferentes causas, podremos orientar de forma más precisa la etiología.
FinanciaciónNo ha sido preciso ningún tipo de financiación para realizar el trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Presentación previa en congresos: Enviado como comunicación oral al 44.° Congreso de la SEEP (Sociedad Española de Endocrinología Pediátrica) bajo el título «Insuficiencia suprarrenal primaria en pediatría». Oviedo. Mayo de 2022.

