


Hemos leído con especial interés el caso clínico publicado de una paciente de 3 años de edad con lesiones cutáneas compatibles con dermatomiositis (DM) sin presencia de debilidad muscular y catalogada como DM juvenil (DMJ) amiopática1. Recientemente, hemos tenido un caso similar de una paciente con lesiones cutáneas típicas, sin otra clínica asociada y sin alteraciones en la analítica sanguínea, electromiograma (EMG) ni resonancia magnética (RM) en el que la biopsia muscular permitió confirmar la inflamación a dicho nivel.
Paciente de 3 años de edad, sin antecedentes médicos personales ni familiares de interés, con lesiones cutáneas eritematosas en cara, manos y superficies de extensión de las extremidades de año y medio de evolución, valoradas y diagnosticadas inicialmente como dermatitis atópica. Clínicamente asintomática desde el punto de vista muscular. En la analítica sanguínea realizada, los reactantes de fase aguda (velocidad de sedimentación globular y proteína C reactiva), enzimas musculares (creatinkinasa, lactatodeshidrogenasa, aldolasa, aspartato aminotransferasa y alanina amnotransferasa) y el factor de von Willebrand presentaban valores dentro de la normalidad. El estudio de autoinmunidad mostró positividad para los anticuerpos antinucleares (título 1/320) con anti-DNAds, anti-Ro, anti-La, anti-Sm, anti-U1RNP y anti-Jo negativos. El EMG fue normal y la RM de cinturas no evidenció signos de edema muscular ni afectación de grasa ni tejido celular subcutáneo. La biopsia cutánea mostró una dermatitis crónica superficial, con degeneración vacuolar de la membrana basal sin vasculitis compatible con DM y la biopsia muscular, fibras musculares sin variabilidad en el tamaño, con agregados focales de linfocitos endomisiales y macrófagos, sin fibras necróticas, atrofia perifascicular ni vasculitis. La tinción inmunohistoquímica para el complejo mayor de histocompatibilidad (HLA) tipo i fue positiva en el sarcolema del 90% de las fibras y, ocasionalmente, en el sarcoplasma (fig. 1). Se diagnosticó de DMJ clínicamente amiopática y se inició tratamiento con bolos de corticoides por vía intravenosa (30mg/kg/día) y posteriormente por vía oral (prednisona 0,5mg/kg/día), hidroxicloroquina (6mg/kg/día), metotrexato subcutáneo (12,5mg/m2/semana) y suplementos de ácido fólico. Dada la falta de mejoría de las lesiones cutáneas, se añadió tratamiento con inmunoglobulinas por vía intravenosa (2g/kg/mes), con mejoría progresiva.
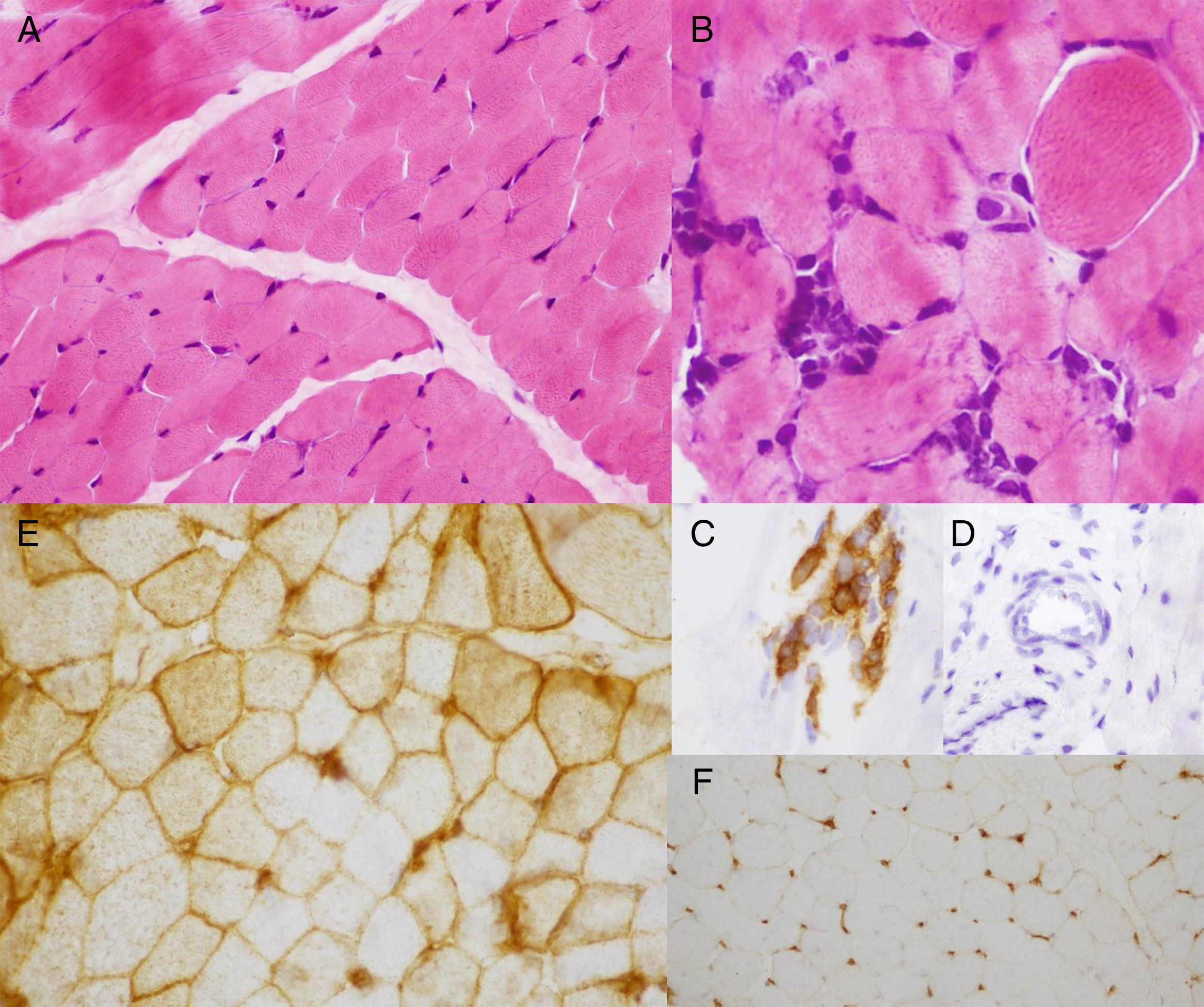
Biopsia muscular con tinción de hematoxilina-eosina con fibras musculares sin variabilidad en el tamaño de las fibras (A) y agregados de células linfohistiocitarias focales (B), que expresan marcadores inmunohistoquímicos para linfocitos maduros (C); sin evidencia de vasculitis (D). Expresión inmunohistoquímica para el complejo mayor de histocompatiblidad tipo i (HLA) en el sarcolema de la práctica totalidad de las fibras y ocasionalmente del sarcoplasma (E). Tejido muscular control (F).
La DMJ es la miopatía inflamatoria más frecuente en la edad pediátrica, una vasculopatía sistémica que afecta a la piel y el músculo esquelético, pero que también puede afectar al tracto gastrointestinal y otros órganos. Su diagnóstico se basa en los criterios de Bohan y Peter2,3, y el objetivo del tratamiento consiste en el control de la enfermedad cutánea y muscular junto con la prevención de complicaciones como la calcinosis, más frecuente en niños que en adultos. Un tratamiento intensivo precoz disminuye la actividad y mejora el pronóstico a largo plazo. La DMJ amiopática es una variante poco frecuente que se caracteriza por la presencia de lesiones cutáneas de DM durante al menos 6 meses, sin evidencia de debilidad muscular. Dalakas y Hohlfeld incluyeron en el subgrupo de pacientes con DM amiopática a aquellos que presentan lesiones cutáneas de DM con ausencia de debilidad muscular en la exploración, permitiendo la presencia de elevación de enzimas musculares hasta 10 veces el valor normal, alteración miopática o inespecífica del electromiograma y/o cambios no específicos o diagnósticos de DM en la biopsia muscular4. En la actualidad, se considera amiopático al paciente que no presenta debilidad muscular en la exploración física ni afectación muscular en las pruebas complementarias y clínicamente amiopático o hipomiopático si la exploración física es normal pero las pruebas complementarias muestran signos de afectación muscular5. La afectación muscular la podemos valorar mediante EMG, RM y/o biopsia muscular. Esta última permite valorar la presencia de infiltrado inflamatorio linfohistiocitario, atrofia de predominio perifascicular y necrosis de las fibras musculares, así como estudiar la sobreexpresión del HLA tipo i en el sarcolema y sarcoplasma de la célula muscular, siendo la que establece el diagnóstico definitivo. En condiciones normales, no existe expresión del HLA tipo i a este nivel, pero en situaciones de activación del sistema inmunológico existe una sobreexpresión generalizada, no limitada a las zonas afectadas, que es característica de las miopatías inflamatorias y que ayuda a diferenciarla de las distrofias musculares, en las que puede existir sobreexpresión pero de menor grado. Además, esta sobreexpresión está presente antes de que exista infiltrado inflamatorio en el músculo6 y no se modifica con el tratamiento inmunosupresor ni en la fase crónica de la enfermedad7.
La calcinosis es una de las complicaciones más graves de la DMJ y su aparición se ha relacionado con un insuficiente control de la enfermedad8,9. En nuestra opinión, realizar una biopsia muscular a todas las DMJ permitiría una mejor clasificación de los pacientes, con implicaciones en el tratamiento y pronóstico a largo plazo.
