


Los modelos de estimación del riesgo de muerte súbita (MS) en miocardiopatía hipertrófica (MCH) pediátrica utilizados en nuestro medio no contemplan algunos parámetros de la práctica clínica habitual. El objetivo fue identificar factores de riesgo no clásicos y evaluar su valor pronóstico.
Pacientes y métodosEstudio observacional retrospectivo, incluyendo pacientes con MCH aislada de 0-18 años, evaluando variables clínicas, genéticas y de imagen. Se estimó el riesgo de MS o evento cardiaco arrítmico mayor (ECAM) según los 3 modelos europeos más empleados, HCM Risk-SCD, algoritmo de la European Society of Cardiology (ESC) y HCM Risk-Kids, analizando su capacidad predictiva al añadir el genotipo y parámetros de imagen cardíaca avanzada.
ResultadosSe incluyeron 77 pacientes, con un seguimiento de 5,25 años. Diez (13%) sufrieron un ECAM. La deformación miocárdica y el genotipo positivo mostraron asociación significativa con los ECAM, y tendencia la presencia de realce tardío (RT) en la resonancia cardiaca (p=0,062). Ocurrieron más eventos (HR=18,5, p=0,006) y más precoces (p=0,022) con variantes en genes distintos a MYBPC3. La inclusión de «genotipo distinto a MYBPC3» y «presencia de RT» mejoró la capacidad predictiva de los modelos en las categorías de riesgo alto (estadístico-C 0,94 vs. 0,84 en HCM-Risk-SCD; 0,88 vs. 0,74 en algoritmo ESC; 0,90 vs. 0,80 en HCM-Risk-Kids) e intermedio (estadístico-C 0,88 vs. 0,51 HCM-Risk-SCD; 0,85 vs. 0,64 guía ESC; 0,84 vs. 0,51 HCM-Risk-Kids).
ConclusionesLa capacidad predictiva de los modelos de riesgo europeos mejora al incorporar las variables «genotipo distinto a MYBPC3» y «presencia de RT», aunque se requieren estudios más amplios que permitan validar su potencial pronóstico.
Models for estimating the risk of sudden cardiac death (SCD) in pediatric hypertrophic cardiomyopathy (HCM) used in our setting do not consider some parameters of routine clinical practice. The objective was to identify non-classical risk factors and evaluate their prognostic value.
Patients and methodsRetrospective observational study, including patients with isolated HCM 0-18 years old, evaluating clinical, genetic, and imaging variables. The risk of SCD or major arrhythmic cardiac events (MACE) was estimated according to the three most widely used European models (HCM Risk-SCD, European Society of Cardiology [ESC] algorithm, and HCM Risk-Kids), analyzing their predictive capacity by adding genotyping and advanced cardiac imaging parameters.
ResultsThe sample included 77 patients followed up for 5.25 years. Ten (13%) experienced a MACE. We found that MACE was significantly associated with myocardial deformation and positive genotype status, and associated, although not significantly, to late gadolinium enhancement (LGE) in cardiac MRI (P=.062). Events were more frequent (hazard ratio=18.5; P=.006) and occurred earlier (P=.022) in association with variants in genes other than MYBPC3. The inclusion of “genotype other than MYBPC3” and “presence of LGE” improved the predictive capacity of the models for the high-risk (C-statistic 0.94 vs 0.84 with HCM Risk-SCD; 0.88 vs 0.74 with ESC algorithm; 0.90 vs 0.80 with HCM Risk-Kids) and intermediate-risk categories (C-statistic 0.88 vs 0.51 with HCM Risk-SCD; 0.85 vs 0.64 with ESC algorithm; 0.84 vs 0.51 with HCM Risk-Kids).
ConclusionsThe predictive capacity of European risk models improves by incorporating the variables “genotype other than MYBPC3” and “presence of LGE”, although larger studies are required to validate their prognostic value.

La miocardiopatía hipertrófica (MCH) es la miocardiopatía familiar más frecuente globalmente y la segunda más prevalente en edad pediátrica1. Se debe en su mayoría a variantes causales en genes sarcoméricos (40-60%), siendo MYBPC3 y MYH7 los más frecuentes2,3. La muerte súbita (MS) es la primera causa de exitus durante la niñez y adolescencia (incidencia 0,8-2% anual), más frecuente en MCH aislada que en el contexto de otro síndrome/enfermedad4–6.
La única medida preventiva eficaz es la implantación de un desfibrilador automático (DAI), con clara indicación tras una taquicardia ventricular (TV) sostenida o una MS recuperada. En prevención primaria, las variables de riesgo arrítmico en MCH pediátrica han sido tradicionalmente extrapoladas de adultos. No obstante, en la última década se han desarrollado modelos predictivos para la estimación del riesgo de MS en MCH aislada aplicables a niños/adolescentes7–10. Los modelos HCM Risk-SCD11, el algoritmo de la guía de la European Society of Cardiology (ESC) de MCH12 y el modelo de riesgo para población pediátrica HCM Risk-Kids13, estiman la probabilidad individual de sufrir una MS en 5 años, teniendo en cuenta variables consideradas clásicamente como la edad, el grosor máximo de la pared del ventrículo izquierdo (GMPVI) o la hipertrofia de ventrículo izquierdo grave, el diámetro de la aurícula izquierda, el gradiente máximo en el tracto de salida del VI (TSVI), la historia familiar de MS, la taquicardia ventricular no sostenida (TVNS) y el síncope inexplicado. Tanto el algoritmo de la guía de la ESC como HCM Risk-Kids han sido validados en menores de 16 años, mientras que HCM Risk-SCD ha sido diseñado para mayores de 16 años10,14,15. Los 3 modelos permiten clasificar a los pacientes en 3 categorías de riesgo de MS: bajo (no indicado el implante de DAI), intermedio (individualizar) o alto (recomendado implante de DAI)11–13.
En los últimos años se ha extendido la utilización de técnicas de imagen avanzada para valorar a pacientes pediátricos con MCH, y evidencias recientes sugieren que algunos parámetros como la presencia de fibrosis miocárdica detectada como realce tardío (RT) extenso en la resonancia magnética cardiaca (RMNc) o parámetros de deformación miocárdica (Strain Longitudinal Global [SLG]) podrían tener valor pronóstico añadido16,17. Por otro lado, el estudio genético en niños con MCH es una práctica clínica cada vez más habitual. Un genotipo positivo permite confirmar la enfermedad, identificar fenocopias de MCH con tratamiento específico y valorar a los familiares18,19. Sin embargo, las implicaciones pronósticas de identificar una variante genética causal en MCH pediátrica no están del todo aclaradas y la información referente al genotipo no está incluida en los modelos de riesgo utilizados en nuestro medio.
Bajo la hipótesis de que la precisión de los modelos de riesgo actuales puede optimizarse y que la estimación del riesgo en población pediátrica es condicional al modelo utilizado, el presente estudio tiene como objetivo evaluar si variables de riesgo no clásicas tienen valor predictivo añadido a los modelos de cálculo de riesgo actuales, con especial énfasis en el genotipo y la presencia de fibrosis miocárdica.
Pacientes y métodosSe realizó un estudio analítico observacional longitudinal retrospectivo de pacientes con MCH aislada (Z score del GMPVI>2) diagnosticada en menores de 18 años en seguimiento en un centro de referencia de cardiopatías familiares, entre febrero de 2009 y agosto de 2022.
Se recogieron variables basales demográficas, clínicas, ecocardiográficas y genéticas, así como parámetros de ergoespirometría y la presencia de RT por RMNc si la información estaba disponible. Se añadió además el valor de la deformación miocárdica (SLG) si se había realizado.
Los estudios genéticos se efectuaron con paneles de al menos 83 genes primarios y secundarios de MCH y, a partir de 2019, con exoma clínico. La clasificación de la patogenicidad de las variantes siguió las recomendaciones del American College of Medical Genetics20. Se consideró un genotipo positivo el hallazgo de al menos una variante genética relacionada con la enfermedad con relevancia clínica (variantes patogénicas, probablemente patogénicas y variantes de significado incierto con muy alta sospecha de patogenicidad).
Se estimó el riesgo de MS según los modelos de riesgo mencionados: el algoritmo de la Guía de la ESC y HCM Risk-Kids en todos los pacientes, y el modelo HCM Risk-SCD en pacientes≥15 años y/o peso≥55kg.
Durante el seguimiento se registraron los casos con implante de DAI en prevención primaria y se recogió la aparición de MS o un evento cardiaco arrítmico mayor (ECAM): MS recuperada, terapia apropiada de DAI o TV con compromiso hemodinámico.
Se estudió la asociación entre la incidencia de MS/ECAM y los factores de riesgo clásicos, y también las variables adicionales genotipo positivo, SLG y RT (presencia y porcentaje de espesor miocárdico afecto). Se valoró la capacidad predictiva de los 3 modelos en la cohorte, así como al incorporar las variables adicionales a los mismos.
El análisis estadístico se realizó con R (versión 4.2.3). Para el cálculo de la asociación entre variables cuantitativas se utilizó la prueba de los rangos de Wilcoxon y para variables cualitativas la prueba Chi-cuadrado de Pearson o prueba exacta de Fisher. Los estudios de supervivencia se analizaron mediante curvas de Kaplan-Meier. Se utilizó la regresión de Cox para modelizar el riesgo a lo largo del tiempo en función de las variables, utilizando como coeficiente de medida del riesgo relativo el hazard ratio (HR), y como medida de bondad de ajuste el estadístico-C a 5 años. Para todos los contrastes de hipótesis se consideró estadísticamente significativo una p<0,05.
Todos los pacientes o responsables legales firmaron un documento informado para la recogida de datos clínicos y genéticos aprobado por el comité de ética del centro (CEIm 19/2019) y de acuerdo con los principios de la Declaración de Helsinki.
ResultadosCaracterísticas basales de la cohorte e incidencia de muerte súbita o evento cardiaco arrítmico mayorSe identificaron 77 pacientes, con edad mediana al diagnóstico 9 [3-12] años, la mayoría (90%) asintomáticos en la valoración inicial. El 62% (45/73) tenía genotipo positivo. Un 46% de los pacientes presentaba una variante en MYBPC3 (21) y un 27% en MYH7 (12). El resto de casos tenían variantes en TPM1 (5; 11%), TNNT2/TNNI3 (3; 7%) y otros genes (4; 9%, FHL1, ALPK3 y ACTC1) (tabla 1 del material suplementario). Ningún caso fue homocigoto para una variante. Un paciente presentó 2 variantes relevantes, en MYH7 y MYBPC3, de alto riesgo por las 3 calculadoras, con implante de DAI y sin ECAM durante el seguimiento.
Los pacientes con genotipo positivo no presentaron diferencias estadísticamente significativas en cuanto a las variables clínicas, ecocardiográficas, ni la presencia de fibrosis miocárdica al diagnóstico con respecto a aquellos con estudio genético negativo (tabla 2 del material suplementario). Durante el seguimiento se implantó un DAI a 27 pacientes (35%), 3 en prevención secundaria y 24 en primaria. Estos últimos presentaban todos un riesgo intermedio-alto según los modelos aplicados.
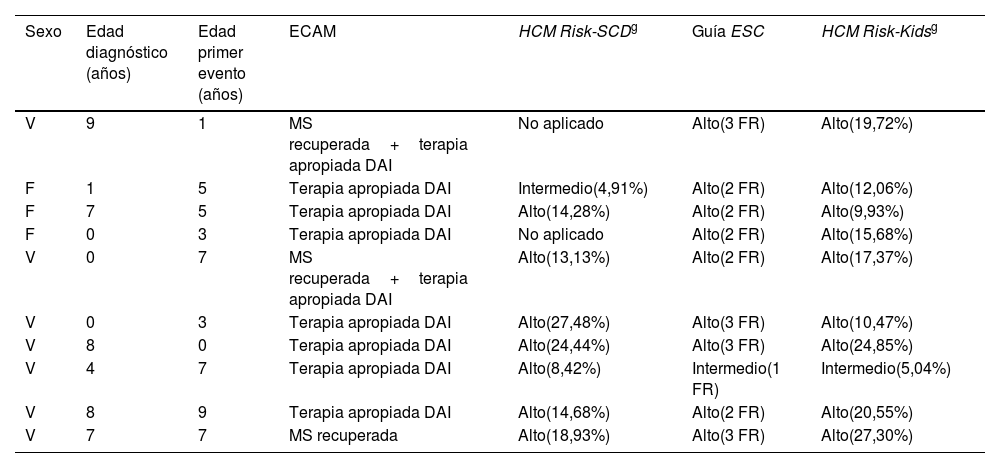
En el tiempo de seguimiento, mediana de 5,25 [4,00-5,50] años, 10 pacientes (13%) presentaron un ECAM (uno MS recuperada, 7 casos terapias apropiadas del DAI y 2 MS recuperada+terapia apropiada de DAI). La tasa de ECAM fue de 2,6 casos (IC 95%: 0,6-8,8) por 100 años-paciente, con una mediana de edad de presentación del primer evento de 13,4 [10,3-15,6] años. La mediana de tiempo libre de evento desde el inicio del seguimiento fue de 2,5 años. No se registró ningún exitus. La tabla 1 muestra los 10 pacientes que experimentaron un evento y la clasificación del riesgo arrítmico en el momento basal según los 3 modelos valorados.
Descripción y valoración del riesgo arrítmico basal de los 10 pacientes que experimentaron un evento cardiaco arrítmico mayor
| Sexo | Edad diagnóstico (años) | Edad primer evento (años) | ECAM | HCM Risk-SCDg | Guía ESC | HCM Risk-Kidsg |
|---|---|---|---|---|---|---|
| V | 9 | 1 | MS recuperada+terapia apropiada DAI | No aplicado | Alto(3 FR) | Alto(19,72%) |
| F | 1 | 5 | Terapia apropiada DAI | Intermedio(4,91%) | Alto(2 FR) | Alto(12,06%) |
| F | 7 | 5 | Terapia apropiada DAI | Alto(14,28%) | Alto(2 FR) | Alto(9,93%) |
| F | 0 | 3 | Terapia apropiada DAI | No aplicado | Alto(2 FR) | Alto(15,68%) |
| V | 0 | 7 | MS recuperada+terapia apropiada DAI | Alto(13,13%) | Alto(2 FR) | Alto(17,37%) |
| V | 0 | 3 | Terapia apropiada DAI | Alto(27,48%) | Alto(3 FR) | Alto(10,47%) |
| V | 8 | 0 | Terapia apropiada DAI | Alto(24,44%) | Alto(3 FR) | Alto(24,85%) |
| V | 4 | 7 | Terapia apropiada DAI | Alto(8,42%) | Intermedio(1 FR) | Intermedio(5,04%) |
| V | 8 | 9 | Terapia apropiada DAI | Alto(14,68%) | Alto(2 FR) | Alto(20,55%) |
| V | 7 | 7 | MS recuperada | Alto(18,93%) | Alto(3 FR) | Alto(27,30%) |
DAI: desfibrilador automático implantable; ECAM: evento cardiaco arrítmico mayor; ESC: Sociedad Europea de Cardiología (European Society of Cardiology); FR: factores de riesgo (n°); HCM: miocardiopatía hipertrófica (hypertrophic cardiomyopathy); M: mujer; MS: muerte súbita; SCD: muerte súbita de origen cardiaco (sudden cardiac death); V: varón.
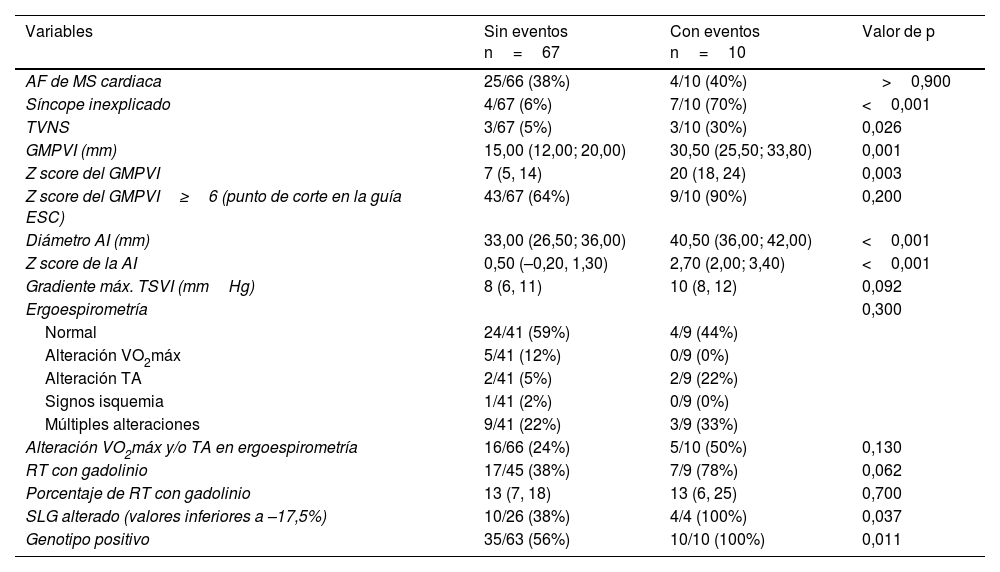
El análisis de asociación de las variables de riesgo clásicas con la aparición de ECAM mostró resultados significativos para síncope inexplicado, TVNS, Z score del GMPVI y Z score de la aurícula izquierda. No se demostró asociación con el gradiente máximo en el TSVI, ni con presentar antecedentes familiares de MS cardiaca, considerados factores de riesgo en MCH en adultos. Por otro lado, la presencia de un valor de SLG anormal y un genotipo positivo presentaron una asociación significativa con la aparición de ECAM. Con respecto a la presencia de RT con gadolinio se observó una tendencia a mayor prevalencia de fibrosis miocárdica entre los pacientes que sufrieron un evento, próxima a la significación estadística (tabla 2).
Variables analizadas en relación con la aparición del evento cardiaco arrítmico mayor
| Variables | Sin eventos n=67 | Con eventos n=10 | Valor de p |
|---|---|---|---|
| AF de MS cardiaca | 25/66 (38%) | 4/10 (40%) | >0,900 |
| Síncope inexplicado | 4/67 (6%) | 7/10 (70%) | <0,001 |
| TVNS | 3/67 (5%) | 3/10 (30%) | 0,026 |
| GMPVI (mm) | 15,00 (12,00; 20,00) | 30,50 (25,50; 33,80) | 0,001 |
| Z score del GMPVI | 7 (5, 14) | 20 (18, 24) | 0,003 |
| Z score del GMPVI≥6 (punto de corte en la guía ESC) | 43/67 (64%) | 9/10 (90%) | 0,200 |
| Diámetro AI (mm) | 33,00 (26,50; 36,00) | 40,50 (36,00; 42,00) | <0,001 |
| Z score de la AI | 0,50 (–0,20, 1,30) | 2,70 (2,00; 3,40) | <0,001 |
| Gradiente máx. TSVI (mmHg) | 8 (6, 11) | 10 (8, 12) | 0,092 |
| Ergoespirometría | 0,300 | ||
| Normal | 24/41 (59%) | 4/9 (44%) | |
| Alteración VO2máx | 5/41 (12%) | 0/9 (0%) | |
| Alteración TA | 2/41 (5%) | 2/9 (22%) | |
| Signos isquemia | 1/41 (2%) | 0/9 (0%) | |
| Múltiples alteraciones | 9/41 (22%) | 3/9 (33%) | |
| Alteración VO2máx y/o TA en ergoespirometría | 16/66 (24%) | 5/10 (50%) | 0,130 |
| RT con gadolinio | 17/45 (38%) | 7/9 (78%) | 0,062 |
| Porcentaje de RT con gadolinio | 13 (7, 18) | 13 (6, 25) | 0,700 |
| SLG alterado (valores inferiores a –17,5%) | 10/26 (38%) | 4/4 (100%) | 0,037 |
| Genotipo positivo | 35/63 (56%) | 10/10 (100%) | 0,011 |
AF: antecedentes familiares; AI: aurícula izquierda; ESC: Sociedad Europea de Cardiología (European Society of Cardiology); GMPVI: gradiente máximo parietal del ventrículo izquierdo; MS: muerte súbita; RT: realce tardío con gadolinio; SLG: Strainlongitudinal global;; TA: tensión arterial; TSVI: tracto de salida del ventrículo izquierdo; TVNS: taquicardia ventricular no sostenida; VO2máx: consumo máximo de oxígeno.
Dada la asociación entre presentar genotipo positivo y la aparición del evento, se evaluó mediante un análisis de Kaplan-Meier la aparición de ECAM en función del tipo de gen afecto. Puesto que MYBPC3 fue el gen más frecuentemente implicado, se compararon los pacientes con genotipo positivo para MYBPC3 frente a aquellos con variantes relevantes en cualquier otro gen. Ocurrieron más eventos (hazard ratio=18,5, IC 95%: 2,31-148; p=0,006) y fueron más precoces en el grupo de pacientes con variantes relevantes en genes distintos a MYBPC3 (fig. 1).
Capacidad predictiva de los modelos de predicción de riesgo.")
Como muestra la tabla 1, ningún paciente que sufrió un ECAM tenía una valoración de riesgo basal baja según los modelos de estimación de riesgo. Se halló una asociación significativa (p≤0,005) entre una mayor puntuación de riesgo y la aparición de ECAM en los 3 modelos analizados, y la probabilidad acumulada de sufrir el evento fue superior en el grupo clasificado de más riesgo para los 3 algoritmos (figura 1 del material suplementario).
Entre los pacientes que fueron clasificados en el grupo de riesgo alto (DAI indicado) al emplear la calculadora HCM Risk-SCD, el 50% presentó un ECAM, el 26% con el algoritmo de la ESC y un 33% entre los de mayor riesgo según HCM Risk-Kids.
Valor predictivo añadido del genotipo y parámetros de imagen avanzada a los modelos predictoresCon los resultados obtenidos de los análisis previos se testó el genotipo, la presencia de RT y el SLG alterado como posibles variables a incluir en un modelo de estimación del riesgo en MCH pediátrica.
El análisis univariante mostró un elevado y significativo (p≤0,02) incremento del riesgo de sufrir un ECAM a lo largo del tiempo si un paciente era clasificado en el grupo de riesgo alto: HR=24,9 con la calculadora HCM Risk-SCD, HR=11,7 según el algoritmo de la ESC y HR=18,8 empleando HCM Risk-Kids.
Por otro lado, los pacientes clasificados en el grupo de riesgo intermedio en las 3 calculadoras, respecto a todos los demás, presentaron un HR<1, es decir, con un teórico menor riesgo de presentar un evento mayor con respecto al resto de pacientes.
El riesgo de ECAM en la cohorte asociado a presentar un genotipo positivo distinto a MYBPC3 fue 19 veces mayor que en los casos con variantes en dicho gen (HR=18,5, p=0,006). Por otro lado, el riesgo de presentar ECAM a lo largo del tiempo atribuible a la presencia de RT mostró un HR=4,2 (p=0,08).
El SLG, aunque se consideró inicialmente, no mostró un incremento de riesgo en el análisis de regresión, por lo que se descartó en los análisis siguientes.
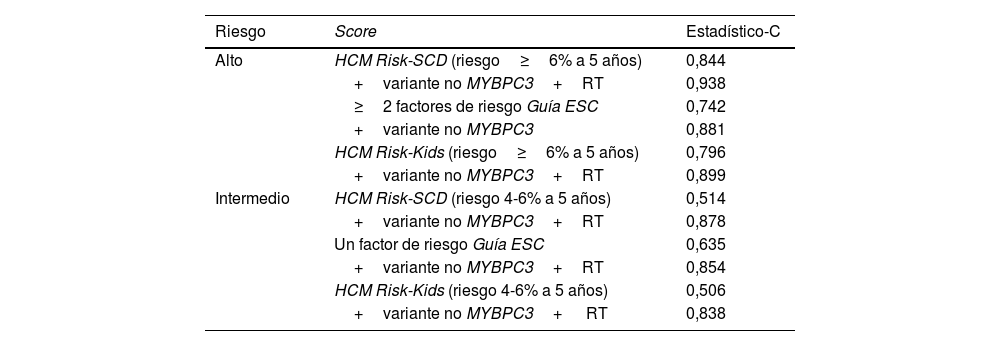
La capacidad predictiva (expresada mediante estadístico-C) de las 2 nuevas variables consideradas relevantes para añadir a los modelos estudiados fue de 0,808 para la presencia de una variante en genes no MYBPC3 y 0,651en el caso de presentar RT.
La tabla 3 muestra, para cada una de las calculadoras, el mejor modelo predictivo (más preciso) que resulta de añadir tanto a la categoría de riesgo alto como a la de intermedio las nuevas variables seleccionadas.
Análisis multivariante del riesgo de MS/ECAM por regresión de Cox en la cohorte. Evaluación de la capacidad predictiva de las categorías de riesgo alto e intermedio según las 3 calculadoras (HCM Risk-SCD, Guía ESC y HCM Risk-Kids) y al añadir las nuevas variables referentes al genotipo y RT
| Riesgo | Score | Estadístico-C |
|---|---|---|
| Alto | HCM Risk-SCD (riesgo≥6% a 5 años) | 0,844 |
| +variante no MYBPC3+RT | 0,938 | |
| ≥2 factores de riesgo Guía ESC | 0,742 | |
| +variante no MYBPC3 | 0,881 | |
| HCM Risk-Kids (riesgo≥6% a 5 años) | 0,796 | |
| +variante no MYBPC3+RT | 0,899 | |
| Intermedio | HCM Risk-SCD (riesgo 4-6% a 5 años) | 0,514 |
| +variante no MYBPC3+RT | 0,878 | |
| Un factor de riesgo Guía ESC | 0,635 | |
| +variante no MYBPC3+RT | 0,854 | |
| HCM Risk-Kids (riesgo 4-6% a 5 años) | 0,506 | |
| +variante no MYBPC3+ RT | 0,838 |
ESC: Sociedad Europea de Cardiología (European Society of Cardiology); HCM: miocardiopatía hipertrófica (hypertrophic cardiomyopathy); RT: realce tardío con gadolinio; SCD: muerte súbita de origen cardiaco (sudden cardiac death).
Para los 3 algoritmos evaluados el comportamiento fue similar. Si bien es cierto que los pacientes clasificados en los grupos de mayor riesgo recibirían un DAI, la capacidad predictiva de cada modelo mejoró aún más (mayor estadístico-C) al añadir la presencia de una variante relevante en genes distintos a MYBPC3 y RT en la resonancia cardiaca. De mayor interés clínico fue hallar que la deficiente capacidad predictiva de las variables independientes correspondientes a pertenecer al grupo de riesgo intermedio mejoró claramente al construir un modelo combinado con las variables presencia de variantes en genes no MYBPC3 y RT.
DiscusiónLa frecuencia acumulada de MS en 5 años tras el diagnóstico de MCH pediátrica aislada se ha descrito entre el 8-10%21. En este estudio la frecuencia de ECAM fue discretamente superior (13%) en un seguimiento de 5,25 años. La tasa de ECAM fue 2,6 casos por 100 años-paciente, ligeramente superior a la registrada por el grupo propulsor del modelo HCM Risk-Kids (1,5-2 casos por 100 años-paciente)10,14. La tasa anual correspondiente de MS/ECAM (en 5 años) fue de 2,0, siendo de 1,5 en el estudio HCM Risk-Kids13. En los pacientes con evento, la mediana de tiempo hasta el mismo fue 2,5 años, y la mediana de edad 13,4 años, datos cercanos a los publicados en el estudio PRIMaCY (mediana de 2,2 años hasta el evento y edad 14,5 años), apoyando posiblemente una etapa de mayor riesgo en la adolescencia22,23.
A pesar de que no existe evidencia bibliográfica clara, el estudio genético parece tener un mayor rendimiento en población infantil respecto a MCH adulta24. Nuestros datos apoyan esta idea; en nuestra cohorte se identifica una variante causal en 6 de cada 10 pacientes testados, lo que podría orientar el estudio familiar y pronóstico25.
Respecto a las técnicas de Strain (valoración de la deformación miocárdica), aunque con un número discreto de casos, los datos sugieren un posible papel pronóstico a validar. Existen aún pocas investigaciones al respecto, pero algunas sugieren una factible correlación entre los marcadores convencionales de MS y este tipo de parámetros de deformación del VI26.
Por otro lado, los pacientes que sufrieron un ECAM, respecto a los que no, presentaron el doble de casos con RT, próximo a la significación estadística. Es posible que esta asociación pudiera confirmarse con mayor número de pacientes, como sugieren estudios más amplios27–29.
Las últimas guías de manejo de miocardiopatías de la ESC (2023) proponen que en los pacientes clasificados de riesgo bajo o intermedio según las calculadoras HCM Risk-SCD o HCM Risk-Kids, la presencia de RT extenso (≥15%) podría utilizarse en la valoración de la indicación de implante profiláctico de DAI30. La Association for European Paediatric and Congenital Cardiology sugiere que, aunque la asociación de RT y riesgo de MS no está del todo definida, ni tampoco establecida la extensión que constituya un mayor riesgo, la presencia de RT extenso podría suponer un factor de riesgo adicional de MS en niños y adolescentes con MCH9. En esta línea, en 2024 se publicó la guía actualizada de la American Heart Association de manejo de MCH, con un apartado para la evaluación del riesgo de MS en menores, proponiendo que pudiera ser útil considerar factores adicionales como la presencia de RT con gadolinio, así como datos de disfunción sistólica31.
Un estudio genético positivo se asoció significativamente con el desarrollo de un ECAM en la cohorte (p=0,011). De hecho, todos los eventos ocurrieron en pacientes con estudio genético positivo. Estos resultados apoyan los descritos en el estudio PRIMaCY, estableciendo un modelo de predicción que, como novedad, incluye la presencia de variantes genéticas patogénicas22.
Los hallazgos de mayor frecuencia y precocidad de eventos en afectos de variantes en genes distintos a MYBPC3 están en línea con investigaciones previas, en las que se asocia la presencia de variantes causales en este gen con fenotipos más leves de comienzo más tardío25. Estos resultados abren la puerta a considerar un papel más notable del genotipo en la estratificación del riesgo25,32,33. Son necesarios estudios más amplios para poder valorar de manera similar el papel pronóstico de variantes en MYH7, y de los casos de dobles variantes.
En la cohorte, los modelos predictivos evaluados parecen predecir bien los eventos, aunque no coinciden exactamente en todos los casos y, además, la mayoría de pacientes clasificados de alto riesgo no sufren eventos mayores en el seguimiento. Por todo ello, parece razonable la necesidad de mejorar y afinar los algoritmos de predicción, donde podría desempeñar un papel relevante la inclusión de nuevas variables.
En semejanza a un estudio sueco reciente en MCH pediátrica, que comparaba la idoneidad a la hora de predecir el riesgo de la calculadora HCM Risk-Kids respecto a asociarse a otras variables (en concreto del ECG), en el presente estudio se realizaron análisis multivariantes para estimar la capacidad predictiva de los diferentes modelos estudiados al añadir las nuevas variables pronósticas propuestas34.
En las 3 calculadoras el grupo de riesgo intermedio mostró un menor riesgo teórico respecto a los demás pacientes (explicable clínicamente puesto que se comparó con el resto, también aquellos de mayor riesgo), con un valor del estadístico-C que traducía una capacidad discriminadora deficiente. Esta capacidad predictiva mejoró significativamente cuando un paciente se clasificaba en el grupo de riesgo intermedio y presentaba además una variante relevante en genes distintos a MYBPC3 y RT en la resonancia. Destaca sobre todo la significación y capacidad predictiva de la variable relativa a la presencia de variantes en genes no MYBPC325.
Las principales limitaciones del estudio son su carácter unicéntrico y el acotado número de eventos (aunque acorde a tratarse de una enfermedad poco frecuente y con casos procedentes de todo el territorio nacional). Desde el punto de vista estadístico (falta de adecuación para el ajuste del modelo), no fue posible incluir en el análisis de regresión el grupo combinado de riesgo intermedio-alto, el de riesgo intermedio respecto al de bajo riesgo, ni el de bajo riesgo frente al resto, por lo que no se pudo analizar la capacidad predictiva que aportarían las nuevas variables en estos grupos.
ConclusionesEn nuestra cohorte de MCH pediátrica, el genotipo positivo y un SLG alterado se asociaron significativamente con la aparición de ECAM. La presencia de RT mostró una tendencia positiva próxima a la significación estadística. Además, hubo más eventos y más precoces en pacientes con variantes genéticas causales en genes distintos a MYBPC3.
Aplicando las herramientas de estimación del riesgo de MS HCM Risk-SCD, la guía de la ESC, y HCM Risk-Kids, tanto en los grupos de riesgo alto como moderado, la capacidad predictiva mejoró al incorporar conjuntamente las variables «genotipo distinto a MYBPC3» y «presencia de realce tardío».
Estos resultados muestran la relevancia de estas variables en la predicción del riesgo arrítmico y apoyan la necesidad de estudios multicéntricos que permitan desarrollar nuevas calculadoras de MS en MCH pediátrica.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesNo existe conflicto de intereses.
Figura suplementaria 1. Capacidad predictiva de los modelos de estimación de riesgo aplicados. A: Relación de los resultados de las clasificaciones HCM Risk-SCD, ESC y HCM Risk-Kids con la aparición de MS/ECAM en el tiempo de seguimiento. B, C, D: Probabilidad acumulada de presentar un ECAM en 5 años por grupo de riesgo según: B) HCM Risk-SCD, C) Guía ESC, D) HCM Risk-Kids.
