





La hipercalcemia hipocalciúrica familiar (FHH) es una de las causas de hipercalcemia; se hereda de forma autosómica dominante, y posee alta penetrancia. Es el resultado de una mutación inactivante en el gen del receptor sensible al calcio (CaSR). Los casos heterocigotos no suelen presentar síntomas y se diagnostican de forma incidental.
Presentamos los casos de tres niñas afectas de una mutación inactivante en heterocigosis, p.Phe789del, en el exón 7 del gen del receptor sensible al calcio (gen CASR) localizado en el cromosoma 3q21 (Ensembl ENSG00000036828). En las muestras sanguíneas se constató hipercalcemia con calcio iónico elevado, hormona paratiroidea normal o elevada, y la calciuria disminuida.
Es importante establecer el diagnostico-diferencial con el hiperparatiroidismo primario. Por lo tanto, en presencia de una hipercalcemia con hormona paratiroidea elevada o normal, se debe realizar el estudio familiar y determinar la calciuria. La aparición de algún miembro afecto en la familia o la aparición de hipocalciuria es suficiente para sospechar esta entidad e indicar el análisis mutacional, para establecer el diagnóstico diferencial con el hiperparatiroidismo primario y evitar tratamientos innecesarios.
Familial hypocalciuric hypercalcemia (FHH) is a cause of hypercalcemia with autosomal dominant pattern of inheritance and high penetrance. In most of the cases it can be shown to be due to an inactivating mutation on the gene coding for the calcium-sensing receptor (CaSR). Heterozygous cases usually do not present symptoms and they are diagnosed as an incidental finding.
We report three affected children with an inactivating heterozygous mutation, p.Phe789del, in exon 7 of the calcium-sensing receptor gene (CASR gene), situated in chromosome 3q21 (Ensembl ENSG00000036828), which results in elevated serum calcium, normal o high level of parathyroid hormone (PTH) and reduced urinary excretion with hypocalciuria.
It is very important to determine the difference between FHH and primary hyperparathyroidism. Therefore, in a mild to moderate PTH-dependent hypercalcemia we must perform a family study and determine the urinary excretion of calcium. The presence of any other affected family member or reduced urinary calcium excretion is enough to suspect FHH, and this should be confirmed by the mutational analysis of the CASR gene, in order to establish the correct diagnosis, differentiated from primary hyperparathyroidism, to avoid unnecessary investigations or operations.
La hipercalcemia hipocalciúrica familiar (FHH) es una de las causas de hipercalcemia; se hereda de forma autosómica dominante, y posee alta penetrancia. Los efectos del calcio sobre la hormona paratiroidea (PTH) y los riñones están mediados por su receptor específico, receptor-sensible al calcio (CaSR), que se encuentra en la membrana de las células de la glándula paratiroides y en las células renales1,2.
La mayoría de los casos de FHH son el resultado de una mutación inactivante en el CaSR, cuyo gen (CASR) se encuentra en el brazo largo del cromosoma 3. Estas mutaciones inactivantes se manifiestan por una hipercalcemia leve o moderada, con PTH normal o elevada y disminución de la excreción de calcio urinario.
PacientesCaso 1Niña de 1 mes, remitida a la consulta para despistaje de FHH. Primera gestación sin incidencias. Parto a término, eutócico. Peso natal (PN): 2.950g. Padres no consanguíneos. Hasta la última revisión realizada a los dos años se ha mantenido asintomática con desarrollo psicomotor y póndero-estatural normales (peso percentil-10 y talla percentil-50).
Caso 2Niña de 2 años y 8 meses. Prima del caso anterior. También acudió a la consulta para despistaje de FHH. Parto a término, eutócico. PN: 3.600g. Padres no consanguíneos. Sus antecedentes personales carecían de interés y presentaba un desarrollo psicomotor y póndero-estatural normales. La última consulta se realizó a los tres años y seis meses manteniéndose asintomática con crecimiento en peso y talla en percentil 90.
Caso 3Niña de 11 años. Prima de las dos anteriores. Parto a término, eutócico. Parto normal. PN: 3.870g. Antecedentes personales sin interés. Desarrollo psicomotor y póndero-estatural normales. Peso percentil-75 y talla percentil-50.
Se realizaron determinaciones en sangre de calcio total, calcio iónico, fósforo, magnesio, PTH, 25-OH-vit. D, 1-25 (OH)2 vit. D. También se determinó la creatinina (Cr), sodio, potasio y cloro. En orina se analizaron el calcio y la creatinina.
En todas las muestras se constató hipercalcemia con calcio iónico elevado, PTH normal o elevada y la calciuria disminuida. Los valores analíticos de las niñas, según la edad, se reseñan en la tabla 1.
Valores analíticos de las tres niñas afectas de mutación.
| Edad (años) | Ca total (mg/dl) | Ca iónico (mMol/l) | PTH (pg/ml) | 25-OH vit D (ng/ml) | 1,25-(OH)2 vit D (pg/ml) | Cr P (mg/dl) | P (mg/dl) | Mg (mg/dl) | IECa (mg/100ml)GFR |
| 9,05–10,4 | 1,12–1,32 | 10–65 | 25–35 | 20–80 | 0,3–0,6 | 3,5–5,2 | 1,7–2,4 | 0,03–0,06 | |
| 1 ½ meses | 12 | 1,6 | 30,6 | 35,7 | 80 | 0,24 | 5,8 | 2,3 | 0,002 |
| 5 meses | 11,6 | 1,5 | 40,3 | 26 | 63 | 0,2 | 5,2 | 2,4 | 0,002 |
| 10 meses | 12,1 | 1,45 | 36,7 | 22,2 | 64 | 0,25 | 4,6 | 2,5 | 0,001 |
| 2 años y 8 meses | 12,1 | 1,38 | 16,5 | 19,8 | 55 | 0,36 | 3,7 | 2,4 | 0,0005 |
| 2 años y 10 meses | 11,1 | 1,38 | 38,5 | 18,6 | 37 | 0,38 | 3,9 | 2,4 | 0,0009 |
| 3 años y 6 meses | 11,4 | – | 34,6 | 20,2 | 34 | 0,3 | 4,1 | 2,4 | 0,0004 |
| 11 años | 11,1 | 1,45 | 54,7 | 14,9 | 48 | 0,45 | 4,1 | 2,3 | 0, 0007 |
25-(OH) vit D: 25 hidroxi vitamina D sérica; 1,25- (OH)2 vit D: 1,25 dihidroxi vitamina D sérica; Ca: calcio; Cr P: creatinina plasmática; IECa: índice de excreción urinaria de calcio; Mg: magnesio plasmático; P: fósforo plasmático; PTH: hormona paratiroidea.
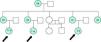
El estudio genético inicial se realizó en la abuela de las niñas (66 años) tras el hallazgo de hipercalcemia durante una evaluación de rutina por osteoporosis. Se estudió molecularmente a la abuela, a sus cinco hijas y a sus tres nietas. Todas, menos una de las hijas, presentaban a nivel del ADN codificante (cADN) cuya nomenclatura sería c.2367_2369 del CTT, la mutación p.Phe789del en heterocigosis (fig. 1). Los datos analíticos se resumen en la tabla 2.
Valores analíticos de los familiares afectos adultos.
| Edad (años) | Ca total (mg/dl) | Ca iónico (mMol/l) | PTH (pg/ml) | 25-OH vit D (ng/ml) | 1,25-(OH)2 vit D (pg/ml) | Cr P (mg/dl) | P (mg/dl) | Mg (mg/dl) | IECa (mg/100ml)GFR |
| 9,05– | 1,12–1,32 | 10,7–38,4 | 25–35 | 20–80 | 0,7–1,1 | 3,1–5,1 | 1,7–2,4 | 0,10–0,09 | |
| 66 | 11 | – | 66,1 | 13 | – | 0,85 | 2,7 | – | – |
| 37 | 10,5 | – | 66 | 18,4 | – | 0,78 | 1,9 | – | 0,003 |
| 35 | 10,7 | – | 26,9 | 31,2 | – | 0,6 | 2,4 | – | 0,008 |
| 29 | 10,7 | – | 41 | 27,5 | – | 0,64 | 2,9 | – | 0,006 |
| 25 | – | – | 31 | 26,4 | – | – | – | – | – |
25-(OH) vit D: 25 hidroxi vitamina D sérica; 1,25- (OH)2 vit D: 1,25 dihidroxi vitamina D sérica; Ca: calcio; Cr P: creatinina plasmática; IECa: índice de excreción urinaria de calcio; Mg: magnesio plasmático; P: fósforo plasmático; PTH: hormona paratiroidea.
La FHH, también denominada hipercalcemia familiar benigna, es una causa poco frecuente de hipercalcemia, que se produce por una mutación inactivante del gen que codifica para el CaSR.
El CaSR se expresa en varios tejidos, incluyendo las glándulas paratiroides, los riñones, médula ósea, osteoclastos y osteoblastos y en otros órganos. Su principal función es regular el balance del calcio. Un aumento en los niveles de calcio en plasma hace que, detectado por el CaSR, actúe este, disminuyendo la secreción de PTH, con la finalidad de normalizar la calcemia1,2.
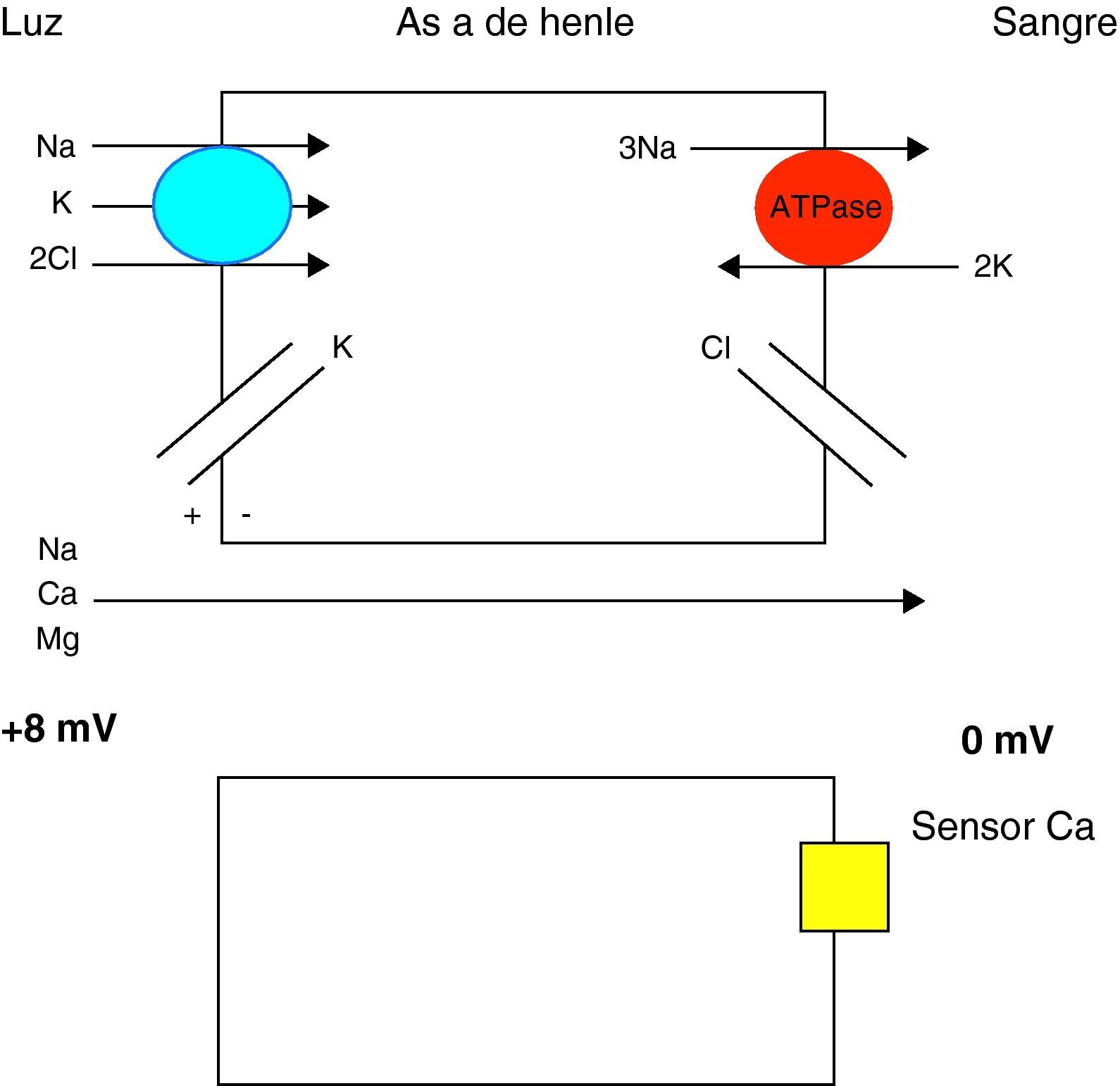
El CaSR es un receptor perteneciente a la familia de los receptores acoplados a proteínas G que se encuentra en la membrana de las células de la glándula paratiroides3. El CaSR es un importante regulador de la excreción urinaria de calcio. El calcio se reabsorbe en el segmento cortical de la rama ascendente de Henle mediante un proceso pasivo. Este mecanismo está regulado por el sensor del calcio, que se encuentra en la superficie basocelular de estas células y detecta los cambios en el calcio iónico sanguíneo (fig. 2). La activación del receptor CaSR, por elevación del Ca iónico en plasma, inhibe los mecanismos de transporte activo de sodio, potasio y cloro y la reabsorción activa de sodio, disminuyendo así la diferencia de potencial transepitelial y, de forma secundaria, la reabsorción pasiva de calcio4,5. Las mutaciones inactivantes en el gen del receptor CaSR provocan un cierto grado de resistencia generalizada al calcio.
Las alteraciones genéticas implicadas con mayor frecuencia en la FHH se encuentran en el gen CASR, localizado en el brazo largo del cromosoma 3. La mayoría de las mutaciones producen un cambio en un único aminoácido que altera la función del receptor6-8. Los pacientes heterocigotos son diagnosticados de forma casual o por cribado de familiares afectados. En familias con FHH y antecedentes de consanguinidad se han descrito formas de hiperparatiroidismo neonatal severo con hipercalcemias severas y trastornos óseos graves cuando se heredan los dos alelos del gen portador de la FHH9. Todos nuestros casos presentaban en heterocigosis una mutación inactivante, p.Phe789del, en el exón 7 del gen del receptor sensible al calcio, localizado en el cromosoma 3q 21.
La escasa expresión clínica pone el punto de interés del diagnóstico de la FHH en la distinción del hiperparatirodismo primario y en evitar tratamientos innecesarios. El perfil bioquímico de hipercalcemia-hipocalciuria se acompaña de tendencia a la hipermagnesemia, niveles séricos inapropiadamente normales o elevados de PTH y metabolitos de la vitamina D10. La ausencia de síntomas de hipercalcemia (debilidad muscular, anorexia, vómitos, estreñimiento, polidipsia-poliuria, pérdida de peso) y el hallazgo de hipercalcemia-hipocalciuria en otros miembros de la familia nos orientan hacia el diagnostico de FHH11. La determinación de la calciuria se entorpece por el problema que supone la recogida de orina de 24 horas en los niños; el cociente o índice Ca/Cr en micción aislada sólo es válido para valorar la hipercalciuria, pero no la hipocalciuria. Por este motivo, se utiliza el índice de excreción de calcio, que se calcula dividiendo el aclaramiento de calcio entre el aclaramiento de creatinina, y se expresan en mg/100ml de GFR12.
El análisis mutacional se debe realizar: a) en los lactantes y niños menores de 10 años, en los cuales el hiperparatiroidismo primario y la FHH son las causas más frecuentes de hipercalcemia cuando la PTH está elevada o normal; b) en los casos atípicos en los que no se demuestra la hipocalciuria, pacientes con fenotipo FHH cuyos padres son normocalcémicos (mutación del CASR de novo); y c) cuando no hay miembros de la familia disponibles para pruebas13.
En resumen, en presencia de una hipercalcemia con PTH elevada o inapropiadamente normal es importante realizar el estudio familiar y determinar la calciuria. La aparición de algún miembro afecto en la familia, o la aparición de hipocalciuria es suficiente para sospechar esta entidad e indicar el análisis mutacional para establecer el diagnóstico diferencial con el hiperparatiroidismo primario y evitar tratamientos innecesarios.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los miembros de la familia, que han aceptado participar en el estudio y comunicar sus resultados.
