




Introducción
El diagnóstico de las epilepsias sigue siendo esencialmente clínico, aunque apoyado por pruebas complementarias cuyos avances técnicos (registro vídeo-EEG [electroencefalograma], estudios de neuroimagen estructural y/o funcional, análisis metabólicos, etc.) facilitan el diagnóstico; y cuya clasificación sindrómica permite presumir cursos evolutivos y eventuales respuestas terapéuticas 1-4. No obstante, los datos publicados sobre la frecuencia relativa de los distintos síndromes epilépticos en la infancia son muy variables, generalmente debido a razones metodológicas 5-10.
La edad representa un factor determinante en la fisiopatología de los distintos tipos de crisis y síndromes epilépticos, ya que las modificaciones estructurales y funcionales que el cerebro va experimentando desde el nacimiento hasta la adolescencia condicionan la expresión clínica y neurofisiológica de las epilepsias. Por tanto, las características epidemiológicas de las distintas epilepsias y síndromes epilépticos infantiles guardarán una estrecha relación con la edad y/o nivel de maduración cerebral 11-14.
El diagnóstico de epilepsia en la edad escolar conlleva la preocupación médica, y especialmente familiar, de la repercusión que la enfermedad pudiera desempeñar en la integración escolar y/o social del niño 15,16, lo que justificaría el análisis de los síndromes epilépticos que se inician en este intervalo de edad.
El objetivo del presente trabajo consiste en analizar las características epidemiológicas y la distribución relativa de los distintos tipos de epilepsias y síndromes epilépticos que tienen lugar entre los alumnos de Educación Primaria (entre 6 y 12 años).
Material y métodos
Se han revisado retrospectivamente las historias clínicas de todos los pacientes con epilepsia cuyo diagnóstico había tenido lugar en edad de cursar Educación Primaria (entre 6 y 12 años) y que acudieron a su control evolutivo ambulatorio en la Unidad de Neuropediatría del Hospital Virgen del Camino de Pamplona entre enero y diciembre del año 2005.
La muestra estaba compuesta por 169 pacientes (85 varones y 84 mujeres). De cada historia clínica se registraron datos epidemiológicos: sexo, edad al diagnóstico, antecedentes personales (embarazo y parto, período neonatal, desarrollo psicomotor y convulsiones febriles), antecedentes familiares de epilepsia y tiempo de evolución; y datos clínicos: tipo de crisis y hallazgos neurológicos (patología asociada), junto a exámenes complementarios: EEG y estudios de neuroimagen (tomografía computarizada [TC] y/o resonancia magnética [RM] craneal) y, en su caso, estudios genéticos, metabólicos y/o neurofisiológicos en orden a establecer la etiología idiopática, sintomática o criptogénica.
Para el diagnóstico y clasificación de las crisis epilépticas y de los síndromes epilépticos se han aplicado los criterios de la International League Againts Epilepsy (ILAE) 4,17. El diagnóstico sindrómico correspondiente a cada paciente fue discutido y consensuado por dos neuropediatras.
Los resultados se expresan como medias y porcentajes con sus intervalos de confianza del 95 % (IC 95 %). Para el análisis estadístico (t de Student, comparación de proporciones) y la representación gráfica se utilizaron los programas informáticos Sigma-Plus (Hardware, 97) y Microsoft Power Point, respectivamente.
Resultados
La edad media de la muestra (n = 169) en el momento del diagnóstico era de 8,8 años (IC 95 %: 8,7-9,0). En la figura 1 se expone la distribución de las edades y sexo en el momento del diagnóstico, objetivándose una prevalencia inversamente relacionada con la edad de diagnóstico. El tiempo medio de seguimiento evolutivo de estos pacientes ha sido de 3,5 años (IC 95 %: 3,28-3,87). La etiología de la epilepsia fue considerada como idiopática en 102 casos (60,4 %), criptogénica en 36 (21,3 %) y sintomática en 31 (18,3 %).
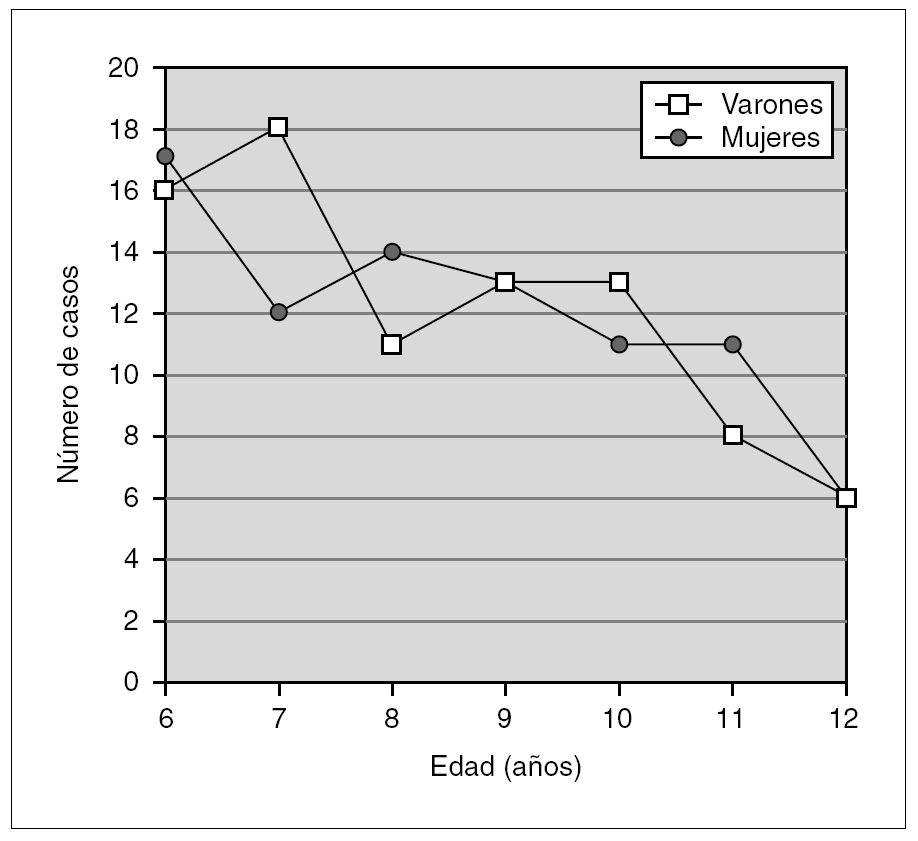
Figura 1. Distribución de la edad y sexo en el momento del diagnóstico.
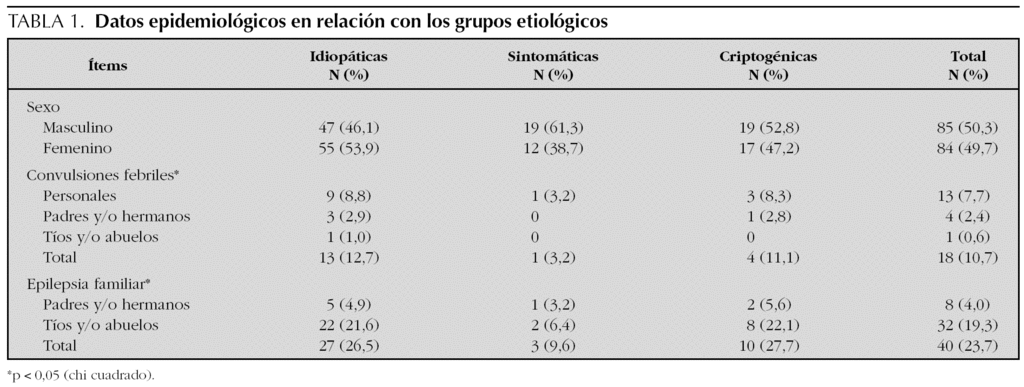
En la tabla 1 se exponen y comparan las características epidemiológicas en relación con los distintos grupos etiológicos. No existían diferencias significativas en la distribución por sexos en ninguno de los grupos etiológicos (50,3 % varones y 49,7 % mujeres). El 10,7 % de los pacientes referían antecedentes personales y/o familiares de convulsiones febriles y el 23,7 % de epilepsia familiar; pero ambos antecedentes se daban preferentemente en las epilepsias idiopáticas y/o criptogénicas.
Como antecedentes personales y/o patología asociada que presentaban estos pacientes en el momento del diagnóstico cabe destacar los siguientes: gran prematuridad (2 casos), hidrocefalia postinfecciosa perinatal (1 caso), patología vascular cerebral perinatal (1 caso), posquirúrgica (1 caso) y postraumática (1 caso), retraso mental severo-moderado (15 casos), déficit motor (7 casos), retraso específico del lenguaje (3 casos), trastorno de atención e hiperactividad (3 casos), hipoacusia neurosensorial (2 casos), trastorno de conducta (1 caso), síndrome de Angelman (1 caso), síndrome de Down (1 caso), síndrome de Rett (1 caso), esclerosis tuberosa (1 caso), síndrome polimalformativo (1 caso) y síndrome del cromosoma 15 invertido y duplicado (1 caso).
Se realizó TC craneal a 84 pacientes (49,7 %), RM craneal a 110 (65 %) y ambas pruebas a 32 pacientes (18,9 %). Es decir, se realizaron pruebas de neuroimagen en el 95,9 % de los pacientes (n = 162), que detectaron alteraciones en 33 casos (20,4 %): encefalomalacia (9 casos), displasia cortical (6 casos), atrofia córtico-subcortical difusa (5 casos) o focal (2 casos), quiste subaracnoideo (3 casos), quiste de septum (1 caso), esclerosis mesial (1 caso), cavernoma (1 caso), múltiples lesiones glióticas en sustancia blanca (1 caso), calcificaciones subependimarias (1 caso), descenso de las amígdalas cerebelosas (1 caso), hidrocefalia tetraventricular (1 caso) y asimetría cortical (1 caso). Los 7 pacientes en los que no se practicaron pruebas de neuroimagen (4,1 %) fueron diagnosticados de epilepsia rolándica (3 casos), ausencias infantiles (2 casos), ausencia juvenil (1 caso) y epilepsia con crisis tónico-clónicas generalizadas del despertar (1 caso).
En la tabla 2 se exponen los tipos de escolarización de los pacientes en relación con los distintos grupos etiológicos. El 66,7 % de los pacientes con epilepsias sintomáticas tenían problemas de aprendizaje, mientras que en las criptogénicas (32,4 %) esta problemática era significativamente menor (p < 0,05); y en las epilepsias idiopáticas (2,9 %) prácticamente no se daban problemas escolares y/o de aprendizaje.

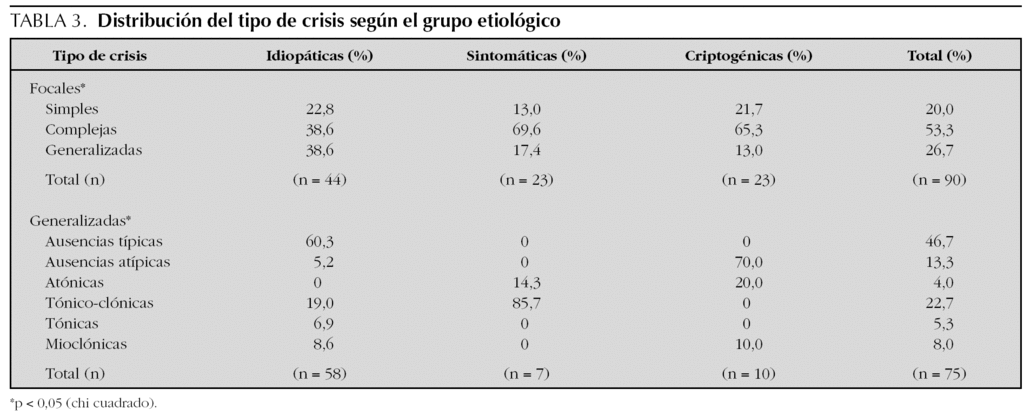
En la tabla 3 se exponen y comparan los distintos tipos de crisis en relación con los distintos grupos etiológicos. Entre las crisis focales, la mayoría de las crisis eran complejas (53,3 %) y, en menor proporción, secundariamente generalizadas (26,7 %) o simples (20 %); no obstante, en el caso de las epilepsias sintomáticas y/o criptogénicas predominaban las crisis complejas sobre las demás. Entre las crisis generalizadas, la mayoría eran ausencias típicas (46,7 %) y, en menor proporción, crisis tónico-clónicas (22,7 %), ausencias atípicas (13,3 %) y crisis mioclónicas (8 %). De los pacientes que presentaron crisis focales o generalizadas, el 86,1 % de los mismos presentaron un único tipo de crisis, el 12,5 % dos tipos distintos y un 1,4 % tres tipos de crisis distintas.
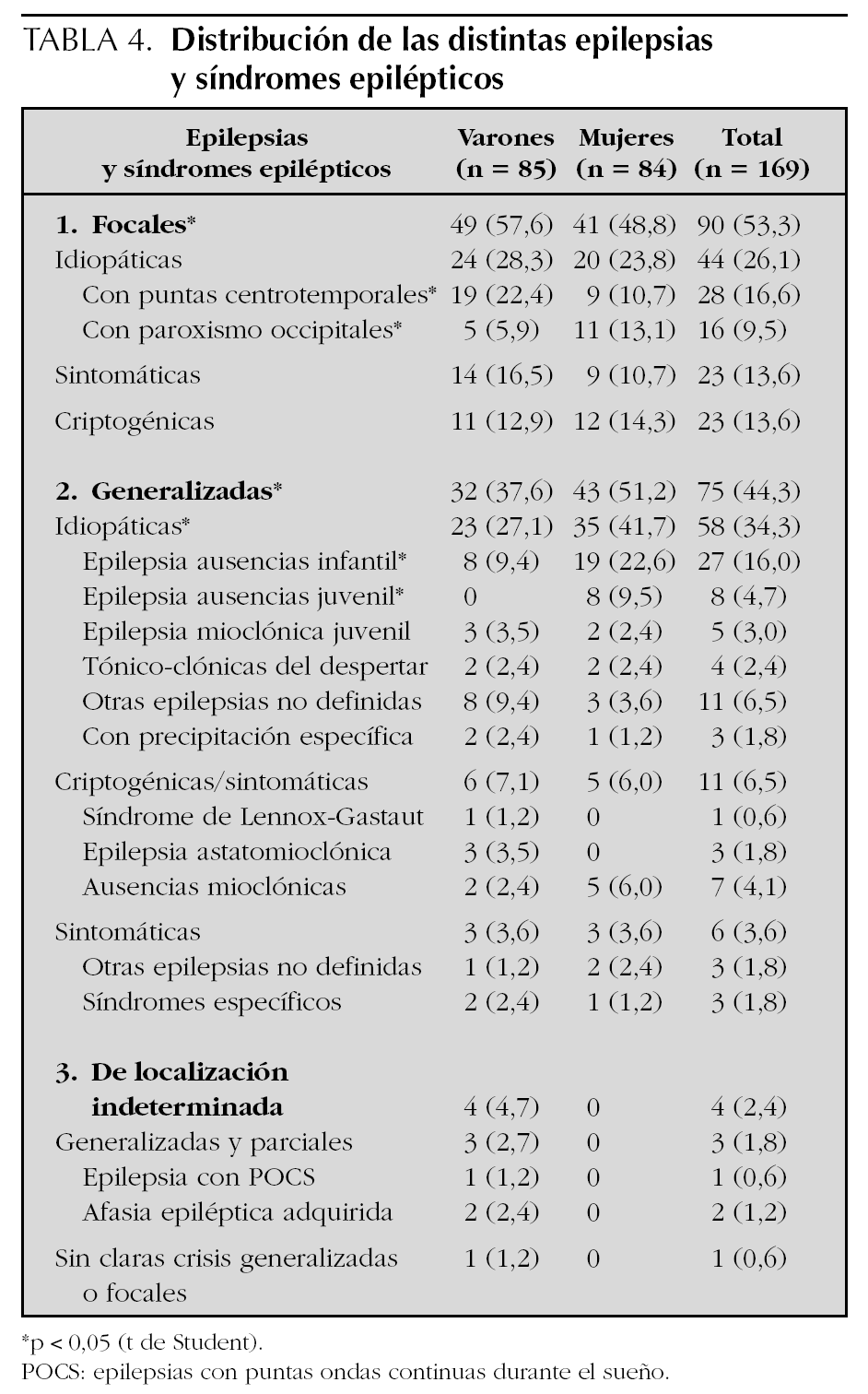
En la tabla 4 se exponen y comparan la distribución de las distintas epilepsias y síndromes epilépticos. El 53,3 % de los pacientes presentaban epilepsias focales, el 44,3 % generalizadas y en el 2,4 % restante eran de localización indeterminada. Entre las epilepsias idiopáticas, el 56,9 % (n = 58) correspondían a epilepsias generalizadas y el 43,1 % restante (n = 44) a epilepsias focales; mientras que entre las epilepsias criptogénicas (63,9 % frente a 27,8 %) y sintomáticas (74,2 % frente a 22,6 %) predominaban las epilepsias focales frente a las epilepsias generalizadas. Las epilepsias y síndromes epilépticos más prevalentes en este intervalo de edad eran de etiología idiopática: epilepsias parciales benignas (26,1 %) y epilepsias ausencias infantil y/o juvenil (20,7 %). En el sexo masculino destacaban las epilepsias rolándicas (22,4 % frente a 10,7 %); mientras que en el sexo femenino lo hacían las epilepsias ausencias (32,1 % frente a 9,4 %).
Discusión
Aunque no existen muchos datos sobre la prevalencia de la epilepsia infantil en España, la extrapolación de los datos publicados 5,18,19 permite calcular que la población seleccionada sería lo suficientemente amplia como para considerarla representativa de nuestro entorno (Censo de población entre 6 y 12 años de edad: 34,248. INE, Navarra); y, además, con posibilidades de obtener resultados que podrían contribuir al conocimiento epidemiológico de la epilepsia infantil.
Las características diferenciales entre los distintos síndromes epilépticos son, a menudo, difíciles de establecer sobre todo al comienzo de la enfermedad y, de hecho, el control evolutivo de estos pacientes suele ser un factor determinante en el diagnóstico definitivo y clasificación sindrómica 20-22. Por tanto, el tiempo medio de seguimiento de la muestra seleccionada de 3,5 años representaría un valor añadido al esfuerzo diagnóstico de la práctica clínica diaria y, en gran medida, permitiría explicar en contraste con otros autores 10,19,23 el hecho de que apenas un 8,9 % de casos fueran catalogados como epilepsias en categorías inespecíficas: 11 casos (6,5 %) de epilepsias generalizadas idiopáticas, 3 casos (1,8 %) de epilepsias generalizadas sintomáticas y un caso (0,6 %) de localización indeterminada. Es más, si aplicáramos el nuevo esquema diagnóstico propuesto por la ILAE que reconoce a la epilepsia tónico-clónica generalizada como una variable fenotípica de epilepsia generalizada idiopática 24, en la población estudiada tan sólo un 4,8 % de casos quedarían clasificados como epilepsias inespecíficas.
A partir de los resultados obtenidos, podría decirse que la mayoría de las epilepsias que se inician durante la Educación Primaria serán idiopáticas (60,4 %), y en menor la proporción criptogénicas (21,3 %) y/o sintomáticas (18,3 %). Entre las epilepsias idiopáticas cabe destacar las epilepsias benignas (26,1 %), especialmente la epilepsia rolándica en varones, y las epilepsias ausencias (20,7 %), especialmente en mujeres. Se trata de 2 síndromes epilépticos bien diferenciados, que conjuntamente representarían el 46,8 % del total de las epilepsias que se inician en este intervalo de edad, cuya determinación genética explicaría la frecuencia de antecedentes familiares encontrados, y que habitualmente no interfieren en la integración escolar y/o social del paciente 4,24-27.
Las epilepsias sintomáticas son secundarias a agresiones cerebrales (síndrome hipóxico-isquémico, traumatismos, accidentes vasculares, etc.), así como a enfermedades estructurales y/o hereditarias (trastornos metabólicos y/o cromosómicos) que con frecuencia cursan con crisis epilépticas 28. Los estudios de neuroimagen y exámenes de laboratorio (análisis metabólicos y/o genéticos) permitieron identificar secuelas debidas a agresiones cerebrales (encefalomacia, atrofia corticosubcortical, etc.) y/o patología subyacente (displasia cortical, esclerosis tuberosa, síndrome de Rett, síndrome de Angelman, encefalopatías metabólicas, etc.). En la edad escolar, la mayoría de las epilepsias sintomáticas serán epilepsias parciales (74,2 %) con una semiología variable en relación con la zona cortical implicada. Su pronóstico dependerá, en gran medida, de la etiología y generalmente se acompañan de una comorbilidad psiconeurológica 29-31 (déficit motor, trastornos perceptivo-sensoriales, retraso mental, etc.) que obliga a que gran parte de estos pacientes requieran apoyo psicopedagógico y/o aulas de apoyo a cargo de equipos especializados (logopedas, fisioterapeutas, psicólogos, etc.) y, en casos concretos, su integración en centros de educación especial, tal y como se constata en esta serie 15,16.
El diagnóstico de las epilepsias criptogénicas es un diagnóstico de exclusión en epilepsias o síndromes epilépticos aparentemente sintomáticos y/o de evolución tórpida, en ausencia de hallazgos patológicos evidentes en los exámenes complementarios. En la edad escolar, la mayoría de las epilepsias criptogénicas serán epilepsias parciales (63,9 %) y, en menor proporción, generalizadas. No obstante, algunos de estos síndromes generalizados tienen antecedentes familiares relativamente frecuentes (epilepsia con ausencias mioclónicas, síndrome de Doose, etc.), su semiología puede ser polimorfa (síndrome de Landau-Kleffner, epilepsia con epilepsias con puntas ondas durante el sueño [POCS], etc.), e incluso presentar refractariedad terapéutica cuya evolución condicionará, en un porcentaje relativamente importante de casos, trastornos cognitivos y/o conductuales que precisarán apoyo psicopedagógico especializado 15,16,31.
En conclusión, en la edad de cursar Educación Primaria la mayoría de las epilepsias que se presentan son de etiología idiopática (epilepsias focales benignas y/o epilepsias ausencias), de pronóstico favorable, y que no suelen interferir en la escolarización del paciente. Las epilepsias sintomáticas y/o criptogénicas son menos frecuentes; sin embargo, en relación con su deterioro psiconeurológico previo y/o eventual refractariedad terapéutica se acompañarán, en un porcentaje importante de casos, de trastornos cognitivos y/o conductuales susceptibles de una intervención psicopedagógica especializada (aulas de apoyo y/o centros de educación especial).
Correspondencia: Dr. T. Durá Travé.
Unidad de Neuropediatría. Hospital Virgen del Camino.
Avda. Pío XII, 10, 8.º C. 31008 Pamplona. España.
Correo electrónico: tduratra@cfnavarra.es
Recibido en junio de 2006.
Aceptado para su publicación en agosto de 2006.
