









Los pacientes con enfermedad neuromuscular constituyen un grupo de riesgo importante para sufrir con frecuencia situaciones de fracaso respiratorio agudo o crónico. Desde que nacen o son diagnosticados requieren un seguimiento por parte del neumopediatra para diagnosticar y tratar las complicaciones respiratorias, que son su principal causa de fallecimiento, dentro de un contexto multidisciplinar.
El soporte ventilatorio y la asistencia a la tos han mejorado la calidad de vida y el pronóstico a largo plazo de muchos de estos pacientes.
En este artículo los autores repasan la fisiopatología, evaluación de la función respiratoria, trastornos del sueño y complicaciones respiratorias más frecuentes en las enfermedades neuromusculares.
En un próximo artículo se analizarán los diversos tratamientos utilizados desde el punto de vista neumológico.
Patients with neuromuscular disease are an important group at risk of frequently suffering acute or chronic respiratory failure, which is their main cause of death. They require follow-up by a pediatric respiratory medicine specialist from birth or diagnosis in order to confirm the diagnosis and treat any respiratory complications within a multidisciplinary context.
The ventilatory support and the cough assistance have improved the quality of life and long-term survival for many of these patients.
In this paper, the authors review the pathophysiology, respiratory function evaluation, sleep disorders, and the most frequent respiratory complications in neuromuscular diseases.
The various treatments used, from a respiratory medicine point of view, will be analyzed in a next paper.
Las enfermedades neuromusculares (ENM) suponen un importante reto para el neumólogo pediátrico. La aplicación de la ventilación no invasiva (VNI) como forma de tratamiento para la insuficiencia respiratoria, tanto aguda como crónica, ha supuesto una mejora fundamental en la calidad de vida y en el pronóstico vital de enfermedades como la distrofia muscular de Duchenne o la atrofia muscular espinal, variando la historia natural de las mismas.
Dentro del seguimiento multidisciplinar que precisan estos pacientes, el neumólogo pediátrico desempeña un papel fundamental cuando la pérdida de fuerza muscular afecta a la musculatura respiratoria o a la de la vía aérea superior. Una evaluación y un seguimiento neumológicos se deben realizar desde el nacimiento o primeros meses de vida (recién nacidos o lactantes con hipotonía), así como desde la confirmación diagnóstica de cualquier ENM que cause una pérdida progresiva de la función respiratoria. El grado de afectación respiratoria no solo va a depender de la enfermedad de base, sino de la aparición de otras complicaciones que puedan conducir con mayor o menor celeridad al fracaso respiratorio crónico, causa más frecuente de morbimortalidad en los pacientes con ENM. Estas complicaciones pueden ser: exacerbaciones respiratorias debidas a infección, apneas del sueño (síndrome de apneas/hipopneas obstructivas del sueño [SAOS]), enfermedad por reflujo gastroesofágico (ERGE), neumonías aspirativas, enfermedad restrictiva pulmonar, etc.
Una reciente guía clínica1 para el manejo respiratorio de estos pacientes resalta la importancia de evaluar en cada consulta médica la salud respiratoria de los mismos, y de detectar la progresión insidiosa de la insuficiencia respiratoria crónica en estos pacientes.
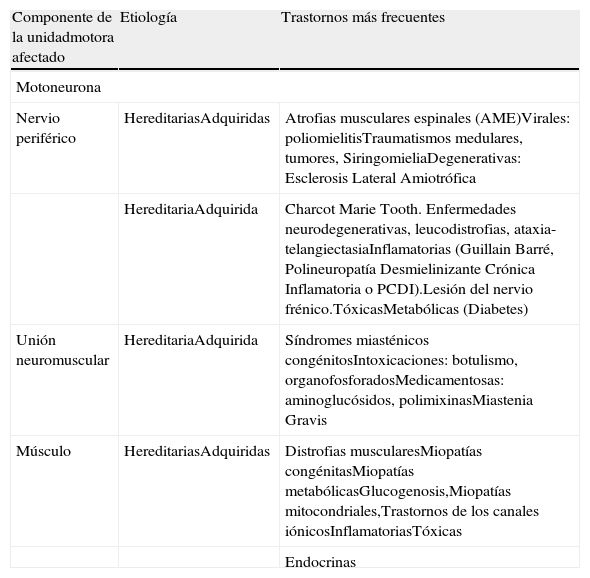
Clasificación de las principales enfermedades neuromuscularesLos trastornos neuromusculares constituyen un grupo de más de 150 enfermedades que afectan a cualquiera de los componentes de la unidad motora, es decir, la unidad funcional constituida por el cuerpo de la motoneurona del asta anterior de la médula espinal, su axón (nervio periférico), la unión neuromuscular y todas las fibras musculares inervadas por esta motoneurona. El efector final de este sistema es el músculo, pero este puede comprometerse en forma primaria o secundaria a la denervación. De acuerdo con este concepto las ENM pueden clasificarse en:
- 1.
Enfermedades de la motoneurona y del nervio (neuropatías).
- 2.
Miopatías o enfermedades primarias del músculo sin alteraciones estructurales en el nervio periférico.
- 3.
Trastornos de la unión neuromuscular.
Cada una de estas afecciones puede ser de causa hereditaria o adquirida, y dentro de estas últimas, producidas por múltiples causas, destacan las de origen inmunológico, las de origen infeccioso (vírico, bacteriano o parasitario), las de origen tóxico-medicamentoso y por último las de origen endocrino-metabólico, dando lugar a la clasificación que se presenta en la tabla 12, que supone una aproximación básica a este tipo de enfermedades, ya que la clasificación de las ENM va cambiando a medida que se conocen nuevos hallazgos sobre las causas de cada una de ellas. En pocos años se ha pasado de una clasificación basada en los rasgos histopatológicos y clínicos a otra en la que los rasgos moleculares son los que articulan la organización de los diferentes grupos, siendo la clínica la que permite establecer subgrupos dentro de un grupo con un trastorno genético común. Actualmente tiene interés la clasificación basada en la biología molecular, lo que permite la creación de nuevos subtipos dentro de un mismo conjunto de síntomas. Esto queda reflejado por ejemplo en la clasificación de las principales miopatías hereditarias (tabla 2).
Clasificación de las enfermedades neuromusculares más frecuentes
| Componente de la unidadmotora afectado | Etiología | Trastornos más frecuentes |
| Motoneurona | ||
| Nervio periférico | HereditariasAdquiridas | Atrofias musculares espinales (AME)Virales: poliomielitisTraumatismos medulares, tumores, SiringomieliaDegenerativas: Esclerosis Lateral Amiotrófica |
| HereditariaAdquirida | Charcot Marie Tooth. Enfermedades neurodegenerativas, leucodistrofias, ataxia- telangiectasiaInflamatorias (Guillain Barré, Polineuropatía Desmielinizante Crónica Inflamatoria o PCDI).Lesión del nervio frénico.TóxicasMetabólicas (Diabetes) | |
| Unión neuromuscular | HereditariaAdquirida | Síndromes miasténicos congénitosIntoxicaciones: botulismo, organofosforadosMedicamentosas: aminoglucósidos, polimixinasMiastenia Gravis |
| Músculo | HereditariasAdquiridas | Distrofias muscularesMiopatías congénitasMiopatías metabólicasGlucogenosis,Miopatías mitocondriales,Trastornos de los canales iónicosInflamatoriasTóxicas |
| Endocrinas | ||
Clasificación de las principales miopatías hereditarias
| 1. Distrofias | ||
| 1.1 Distrofinopatías: | -Duchenne | |
| -Becker | ||
| 1.2 Distrofia de cintura: | * Autosómica dominante | |
| * Autosómica recesiva: | - Sarcoglicanopatías | |
| - Calpainopatías | ||
| - Disferlinopatías | ||
| 1.3 Distrofia fascioescapulohumeral | ||
| 1.4 Distrofia oculofaríngea | ||
| 1.5 Distrofias congénitas: | * Sin afectación SNC | |
| * Con afectación SNC | ||
| 1.6 Otras distrofias: | * Emery-Dreyfuss | |
| * Distales | ||
| 2. Miotonías | ||
| 2.1 Distrofia miotónica de Steinert | ||
| 2.2 Miopatía miotónica proximal | ||
| 2.3 Miotonías no distróficas: | * Parálisis periódicas | |
| - Hipercaliémica/paramiotonía congénita | ||
| - Normocaliémica | ||
| - Hipocaliémica | ||
| * Miotonía congénita | ||
| - Dominante (Thomsen) | ||
| - Recesiva (Becker) | ||
| * Condrodistrofia miotónica (Schwartz-Jampel) | ||
| 3. Congénitas | ||
| 3.1 Central Core/Hipertermia maligna | ||
| 3.2 Dismadurativas | ||
| 3.3 Nemalínica | ||
| 4. Metabólicas | ||
| 4.1 Glucogenosis: Pompe, McArdle | ||
| 4.2 Miopatías lipídicas | ||
| 4.3 Miopatías mitocondriales |
Modificado de: Grupo de Estudio de Enfermedades Neuromusculares.2
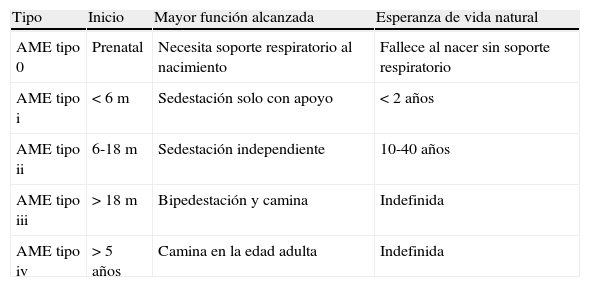
También la clasificación de las atrofias musculares espinales (AME) está evolucionando. Recientemente se tiende a dividir entre AME con mutaciones en el gen SMN1 (5q13, la mayor parte de las formas clásicas), y aquellas con mutaciones en genes distintos al SMN1 (non-5q SMA diseases). Entre estas últimas destaca por su afectación respiratoria la AME con distrés respiratorio 1 (SMARD 1). (tabla 3)3.
Clasificación de las atrofias musculoespinales (AME) 5q13
| Tipo | Inicio | Mayor función alcanzada | Esperanza de vida natural |
| AME tipo 0 | Prenatal | Necesita soporte respiratorio al nacimiento | Fallece al nacer sin soporte respiratorio |
| AME tipo i | <6m | Sedestación solo con apoyo | <2 años |
| AME tipo ii | 6-18m | Sedestación independiente | 10-40 años |
| AME tipo iii | >18m | Bipedestación y camina | Indefinida |
| AME tipo iv | >5años | Camina en la edad adulta | Indefinida |
La distrofia muscular de Duchenne (DMD) es una enfermedad neuromuscular que junto con la distrofia de Becker forma el grupo de las distrofinopatías. Están causadas por mutaciones en el gen de la distrofina y presentan herencia recesiva ligada al cromosoma X, por lo que afectan principalmente al sexo masculino. En la DMD, en concreto, el defecto se localiza en el brazo corto del cromosoma X locus 21 (Xp21). La distrofina es una proteína localizada en la cara citoplasmática de la membrana de las fibras musculares, y su papel es estabilizar las membranas plasmáticas durante la contracción muscular. Cuando la distrofina no está presente la estructura muscular carece de los efectos protectores y organizadores de esta proteína, por lo que la contracción del músculo causa la rotura de las membranas musculares y la consecuente debilidad muscular, que es el síntoma prínceps de la enfermedad3,4.
La incidencia de la enfermedad de Duchenne se estima entre 1 de cada 3.500 a 5.000 varones nacidos vivos. La debilidad muscular es de evolución progresiva y más rápida que en el resto de las distrofias. Afecta inicialmente la cintura pélvica y las extremidades inferiores, y posteriormente la cintura escapular y los miembros superiores. Los cambios histológicos son evidentes desde el nacimiento, pero el comienzo de la clínica suele aparecer entre los 3 y los 5 años.
DiagnósticoLa sospecha diagnóstica aparece con la debilidad muscular y los síntomas característicos. A nivel de laboratorio se observa un aumento de las enzimas musculares (CPK) que puede estar presente desde el nacimiento y que presenta un pico alrededor de los 2 años de vida, con valores entre 10 y 50 veces los normales, posteriormente descienden aunque siempre manteniéndose por encima de la normalidad. El electromiograma revela cambios miopáticos que consisten en potenciales polifásicos pequeños. El estudio genético molecular es el que confirma el diagnóstico en la mayoría de los casos, identificando el tipo específico de mutación del exón o exones afectados.
La biopsia muscular revela necrosis segmentaria y regeneración posterior; cuando el proceso de necrosis-regeneración se agota el músculo es sustituido por grasa y tejido conectivo. Mediante inmunohistoquímica y utilizando anticuerpos antidistrofina se puede evaluar tanto la cantidad como la calidad de la distrofina y/o de las glucoproteínas asociadas a ella. La ausencia completa de la distrofina o cifras de menos de 3% son específicas y características del fenotipo grave de DMD. Sin embargo, la biopsia no suele ser necesaria para el diagnóstico, y suele reservarse para los casos dudosos en los que la clínica no está clara o no existe historia familiar.
Clínica y evoluciónLos primeros síntomas se advierten tempranamente, alrededor de los 3 años de edad. El retraso en la adquisición de la marcha puede ser el primero, apareciendo posteriormente dificultad para caminar, correr, subir y bajar escaleras, así como caídas frecuentes. Tras estas caídas presentan dificultad para levantarse del suelo y lo hacen ayudándose con las manos en una maniobra característica conocida como maniobra de Gowers. La debilidad afecta inicialmente a la cintura pélvica y posteriormente a la escapular y musculatura distal. Destaca la hipotonía y la hiporreflexia osteotendinosa. En la marcha hay aumento de base de sustentación e hiperlordosis y ocasionalmente se realiza en punta de pies. También es característica la pseudohipertrofia de las pantorrillas (2/3 de los pacientes). El deterioro es progresivo y suelen perder la marcha entre los 7 y los 13 años de edad; la pérdida de la marcha es un punto de inflexión en la aparición y progresión de las deformidades esqueléticas.
A nivel respiratorio la debilidad del diafragma, los músculos intercostales y accesorios produce un defecto restrictivo y una insuficiencia respiratoria hipercápnica. La función pulmonar muestra un patrón restrictivo con diminución de la capacidad vital (CV), la capacidad pulmonar total (CPT) y la capacidad residual funcional (CRF), preservando relativamente el volumen espiratorio forzado del primer segundo (FEV1) y el índice FEV1/FVC. El descenso de los volúmenes pulmonares es de un 6-10,7% por año a partir de los 10 años de edad, y el de la CV es de unos 200ml al año5,6.
La disminución de la fuerza inspiratoria, glótica y de la musculatura espiratoria provoca un decremento de los flujos máximos que condiciona la pérdida de la capacidad para toser y el mal manejo de las secreciones bronquiales.
La disminución del esfuerzo respiratorio y del tono de la musculatura de la vía aérea que se produce durante el sueño fisiológico, sobre todo en las fases REM, se ve acentuada en estos pacientes produciendo hipoventilación y síndrome de apneas-hipopneas obstructivas del sueño (SAHOS). Aunque no existe un único parámetro de función pulmonar o gasométrico que pueda predecir con exactitud su aparición, el mejor predictor de hipoventilación nocturna es la FVC, por lo que se recomienda realizar estudios de sueño con valores de FVC inferiores a 60%7. El SAOS suele aparecer durante el final de la primera década (edad media 8 años) y la hipoventilación nocturna a principios de la segunda (edad media 13 años). Estas alteraciones deben detectarse precozmente, ya que suelen preceder a la insuficiencia respiratoria diurna, y además una actuación precoz puede prevenir el deterioro neurocognitivo y las consecuencias cardíacas7.
La hipercapnia diurna es un signo de mal pronóstico e indica una progresión de la enfermedad. El grado de hipercapnia no está directamente relacionado con el grado de debilidad muscular, pues intervienen otros factores descritos como alteraciones de la mecánica del aparato respiratorio, el control de la ventilación, la fatiga muscular, los trastornos respiratorios del sueño y la disfunción de la vía aérea superior8,9.
El inicio electivo del soporte ventilatorio cambia la evolución y mejora el pronóstico de los pacientes, ya que ha demostrado mejorar los gases arteriales, estabilizar las manifestaciones clínicas respiratorias, reducir las necesidades de hospitalización, aumentar el bienestar e incrementar la supervivencia, mejorando sobre todo la calidad de vida como se comentará más adelante10.
La frecuencia de escoliosis es del 95% de los pacientes y es siempre progresiva en el momento en que el paciente pasa a una silla de ruedas. La capacidad pulmonar tiene una relación directa con el grado de deformidad, por lo que cada 10% en la progresión de la curva escoliótica se asocia a una disminución del 4% de la capacidad pulmonar. Los pacientes, a diferencia de las escoliosis idiopáticas, presentan una gran pérdida de la capacidad pulmonar con pocos grados de deformidad de la escoliosis, y aquellos que todavía caminan tienen menos progresión de la deformidad escoliótica, por tanto la rehabilitación con aparatos externos puede favorecer que persista la marcha, ayudar a que no progrese la deformidad y, a su vez, que la capacidad pulmonar no disminuya.
La DMD causa una miocardiopatía dilatada y anomalías en la conducción. Se produce una fibrosis de la pared posterobasal del ventrículo izquierdo, por lo que en el electrocardiograma pueden observarse ondas R altas en las derivaciones precordiales y Q profundas en las derivaciones de los miembros. La afectación cardíaca no suele ser sintomática hasta la adolescencia y/o en los estadios avanzados de la enfermedad, infecciones o intervenciones quirúrgicas donde pueden aparecer arritmias e incluso fallo cardíaco causando la muerte repentina.
La disfagia y los trastornos de aspiración crónica aparecen en las fases avanzadas y comienzan con infecciones respiratorias de repetición y malnutrición. Al igual que en la insuficiencia respiratoria, la detección precoz permite prevenir daños pulmonares crónicos y mejora el pronóstico, así como la calidad de vida. El compromiso de la musculatura lisa también se puede manifestar como trastornos gastrointestinales, entre ellos dilatación gástrica aguda, retrasos en el vaciamiento gástrico y estreñimiento.
El 30% de los pacientes tiene afectación intelectual, con un promedio de pérdida de 20 puntos de IQ comparado con la población control.
La supervivencia ha cambiado en los últimos años, sobre todo con la introducción de la ventilación no invasiva ha aumentado la expectativa de vida de los 15 años a los 40. Los pacientes con insuficiencia respiratoria hipercápnica establecida fallecían antes de los 10 meses y actualmente 2/3 partes sobreviven al menos 5 años más11.
Atrofia muscular espinalSe trata de una enfermedad congénita de herencia autosómica recesiva cuya incidencia se ha estimado entre 7,8-10 por 100.000 nacidos vivos. El defecto genético se localiza en el brazo corto de cromosoma 5; el más común es una deleción homozigota del exón 7 de la Survival Motor Neuron (SMN) locus 5q13. El gen SMN tiene 2 copias en cada cromosoma 5, la SMN1 y la SMN2, que forman una duplicación invertida. El gen SMN1 está siempre alterado y el gen SMN2 está presente en número de 1 a 5 copias en los afectados. Cuantas más copias de SMN2 haya, en general será más benigno el fenotipo, por lo que se considera al gen SMN2 como un modificador fenotípico12. Esta enfermedad es la segunda causa de muerte por enfermedad congénita recesiva después de la fibrosis quística. Se caracteriza principalmente por la debilidad muscular y la atrofia debida a una degeneración celular del asta anterior de la médula espinal y las motoneuronas bulbares inferiores. Se clasifica en 5 tipos según la edad de inicio y la evolución clínica (ver apartado de clasificación [tabla 3]).
DiagnósticoTras la sospecha clínica la confirmación diagnóstica se hace mediante el estudio genético molecular. El análisis del ADN por amplificación selectiva (PCR) de los exones 7 y 8 del gen SMN1 mostrará la ausencia (deleción homocigota) de dicho gen y la presencia del gen SMN2. Este resultado confirma el diagnóstico de AME en casi todos los casos (95%). Los casos restantes pueden deberse a mutaciones puntuales en uno de los alelos y una deleción en el otro. Dicha deleción se ha observado en una amplia gama de fenotipos, desde la grave afectación congénita hasta individuos prácticamente asintomáticos, sin que exista clara correlación entre las deleciones y un fenotipo determinado. En el análisis de sangre lo único reseñable es una discreta elevación de las CPK, aunque sin llegar a los niveles de las distrofinopatías.
El electromiograma muestra signos de denervación, como potenciales de fibrilación espontánea y ondas positivas, así como potenciales de unidad motora de gran amplitud. La velocidad de conducción nerviosa está conservada.
La biopsia muscular muestra un patrón neuropático característico, con fibras hipertrofiadas que normalmente muestran propiedades histoquímicas de fibras tipo i (lentas) y fibras pequeñas de forma redondeada. Al igual que en la DMD no suele ser necesaria excepto en los casos dudosos. En caso de sospecha clínica que no se pueda confirmar con el estudio genético se debe hacer el diagnóstico diferencial que incluye otras entidades (tabla 4)13.
Diagnóstico diferencial de la hipotonía neonatal
| AME con distrés respiratorio (SMARD) (paresia diafragmática) |
| AME ligada al cromosoma X |
| AME con atrofia olivopontocerebelar |
| Artrogriposis múltiple congénita |
| Mielopatía hipóxico-isquémica |
| Mielopatía traumática |
| Síndrome de Zellweger |
| Síndrome de Prader Willi |
| Distrofia miotónica congénita |
| Déficit de citocromo C oxidasa |
| Glucogenosis tipo ii (Pompe) |
| Distrofias musculares congénitas |
El espectro clínico y la evolución son diferentes según el tipo, por lo que lo dividiremos por apartados:
Atrofia medular espinal tipo 0, prenatal o congénitaInicio prenatal con ausencia o disminución de movimientos fetales, debilidad severa al nacimiento con hipotonía profunda y arreflexia. Fallo respiratorio precoz con expectativa de vida inferior a los 6 meses sin soporte ventilatorio.
Atrofia medular espinal tipo i o enfermedad de Werdnig-HoffmanInicio clínico entre el nacimiento y los 6 meses de vida. Presentan debilidad muscular, arreflexia e hipotonía generalizada que afecta más a las piernas que a los brazos, pobre control cefálico y ausencia de sedestación. Son característicos el llanto débil, la tos escasa, la disfagia y mal manejo de las secreciones orales. La afectación de la musculatura intercostal, pero con el diafragma respetado, confiere al tórax un aspecto campaniforme además de causar un tipo de respiración paradójica característica, con abdomen globuloso.
Atrofia medular espinal tipo iiLos síntomas aparecen a partir de los 6 meses y antes de los 18 meses; pueden sentarse sin soporte, aunque nunca llegan a andar. Presentan debilidad progresiva proximal de predominio inferior, hipotonía y arreflexia. Pueden presentar temblor de manos, contracturas y anquilosis mandibular. Desarrollan una escoliosis progresiva que, asociada a la debilidad de la musculatura intercostal, causa la afectación pulmonar restrictiva. La rehabilitación para evitar las contracturas, la cirugía de la escoliosis y la ventilación mecánica no invasiva son pilares básicos en el tratamiento que determinarán la evolución. También es importante el tratamiento de la disfagia y el control nutricional.
Atrofia medular espinal tipo iii o enfermedad de Kugelberg-WelanderLos síntomas se aprecian a partir de los 18 meses. Los pacientes son capaces de caminar en algún momento, aunque la evolución de la enfermedad acaba confinándolos a la mayoría a una silla de ruedas. La afectación respiratoria y la escoliosis son menos graves y pueden no necesitar soporte respiratorio. En algunos casos la enfermedad se estabiliza y los pacientes pueden caminar, aunque con dificultad, durante décadas. La expectativa de vida es similar a la población general.
Atrofia medular espinal tipo ivLa forma tipo iv aparece en la segunda o tercera décadas de la vida y, habitualmente, son pacientes que deambulan durante toda la vida y con una afectación clínica en general leve o moderada14.
Los pacientes con AME tienen un cociente intelectual normal y en algunos casos superior a la media.
La evolución y el pronóstico de la AME dependen obviamente del fenotipo, pero también del manejo del paciente y de la actitud respecto a las medidas terapéuticas15,16. Se recomienda como primer objetivo, una vez confirmado el diagnóstico, realizar un plan de tratamiento consensuado con la familia, determinar los objetivos y los límites, evitando las decisiones en los momentos críticos con el paciente en fallo respiratorio agudo. Idealmente el manejo del paciente con AME debe ser multidisciplinar. Si la familia decide realizar un tratamiento activo el primer paso sería realizar un estudio del sueño para detectar la hipoventilación nocturna, una ventilación no invasiva durante el sueño permitirá a la musculatura descansar, normalizará la respuesta respiratoria a la hipercapnia y permitirá al paciente manejarse mejor durante el día sin ventilación. El uso de la ventilación no invasiva también puede ser muy útil durante las reagudizaciones respiratorias, así como los asistentes de la tos y la fisioterapia respiratoria; estos tratamientos se revisan en un artículo posterior.
Fisiopatología de la disfunción respiratoria de las enfermedades neuromuscularesAunque el grado y la velocidad de instauración de la disfunción pulmonar en los niños con ENM depende de la enfermedad de base, de la coexistencia o no de otros trastornos y de la variabilidad individual, todos comparten unas mismas anomalías fisiológicas que determinan una afectación respiratoria típica17 que lleva a la insuficiencia respiratoria y, ulteriormente, al fracaso respiratorio.
El fracaso respiratorio es la causa más frecuente de morbimortalidad en los pacientes con enfermedad neuromuscular progresiva. Las causas de este fracaso respiratorio se pueden categorizar en aquellas que resultan de enfermedad parenquimatosa pulmonar que condiciona una insuficiencia respiratoria hipoxémica, y en las que se originan de la disfunción de la bomba respiratoria; esta última comprende la musculatura respiratoria, la pared torácica y el centro de control respiratorio18 (fig. 1). Aunque la enfermedad del parénquima pulmonar (neumonías, episodios recurrentes de aspiración…) puede provocar problemas respiratorios en estos niños, el fracaso de la bomba respiratoria suele ser el origen de su fracaso respiratorio crónico.
Disfunción de la bomba respiratoriaLa musculatura respiratoria
La afectación de la musculatura en los pacientes con ENM incluye casi de manera constante tanto a los músculos inspiratorios como a los espiratorios. La debilidad de la musculatura respiratoria y una tos ineficaz son las principales causas de las complicaciones que dan lugar a morbimortalidad. El diafragma es el músculo inspiratorio más importante, responsable aproximadamente del 70% de la ventilación en reposo. La musculatura inspiratoria accesoria incluye los músculos intercostales externos, el escaleno y la musculatura esternocleidomastoidea. Los músculos espiratorios incluyen los intercostales internos y los músculos de la pared abdominal; estos músculos no son necesarios durante la respiración en reposo, por las propiedades elásticas pasivas de la pared torácica, pero son importantes para generar una tos eficaz19. Al igual que ocurre con cualquier otro órgano, existe una reserva sustancial en el sistema respiratorio e, inicialmente, los síntomas respiratorios pueden ser mínimos. Asumiendo que el cerebro y los pulmones funcionan adecuadamente, el fracaso respiratorio no suele ocurrir hasta que la musculatura respiratoria ha perdido hasta un 70-75% de su fuerza. La afectación de la musculatura respiratoria también puede verse enmascarada en estos niños por la disminución de su actividad física, como consecuencia de la debilidad de la musculatura de los miembros. No es infrecuente, por lo tanto, que la debilidad muscular respiratoria pase desapercibida hasta que un episodio agudo, como una infección o una aspiración, ocasionen un fracaso respiratorio.
Inicialmente, esta disminución en la fuerza de la musculatura respiratoria se traduce en una tos ineficaz y en problemas para el manejo de las secreciones de la vía aérea, lo que predispone a que estos pacientes presenten atelectasias de repetición e infecciones respiratorias. Una tos eficaz constituye un mecanismo de defensa del individuo para aclarar la vía aérea. Las fases de la tos se clasifican en: 1) inspiratoria; 2) compresiva; y 3) espiratoria. Durante la fase inspiratoria los sujetos sanos tienen volúmenes previos de alrededor del 85-90% de su capacidad inspiratoria. Después de que se inhale el suficiente volumen de aire, la glotis se cierra automáticamente, lo que previene cualquier salida de aire. La contracción de los músculos espiratorios (fase compresiva) mientras la glotis está abierta inicia la fase de expulsión durante la cual el aire de los pulmones es forzado a salir. Se generan flujos espiratorios muy elevados, que facilitan la eliminación de las secreciones bronquiales. No se puede obtener una tos eficaz si una de estas fases falla, lo que puede ocurrir en pacientes con ENM por varias razones. La debilidad de la musculatura inspiratoria disminuye la capacidad del individuo para realizar una inspiración profunda para dilatar las vías aéreas intratorácicas20. La disminución de la fuerza y velocidad de contracción de los músculos espiratorios provoca una disminución de los flujos máximos, lo que condiciona la disminución del esfuerzo para toser y mal manejo de las secreciones. En condiciones normales, los picos de flujo que se alcanzan así de forma espontánea con la tos oscilan entre 6 y 17l/s. En las ENM se observan valores muy inferiores, que pueden llegar a ser incompatibles con la expectoración21.
La afectación de los músculos inspiratorios produce un patrón restrictivo, con disminución de la CV, la CPT y la CRF, con una relación FEV1/CVF relativamente normal. Si no existe una afectación importante de la musculatura espiratoria los volúmenes residuales se conservan. La caída de esta CRF en los pacientes con ENM está condicionada por la falta de tono de los músculos torácicos, que no ejercen una oposición significativa al retroceso elástico del pulmón; junto a esta debilidad hay disminución de la distensibilidad de la pared torácica. Estos cambios dependen fundamentalmente del aumento de la rigidez de la caja derivada de la disminución mantenida de su expansión y la subsiguiente rigidez de las articulaciones. Las alteraciones de los volúmenes pulmonares son atribuibles a una combinación de debilidad muscular con alteraciones de las propiedades mecánicas de los pulmones y la pared torácica.
La reducción de la CRF aumenta el trabajo respiratorio19 dado que, a menores volúmenes pulmonares, la distensibilidad pulmonar disminuye. Además, la reducción en los volúmenes pulmonares por la disminución de la capacidad inspiratoria favorece el colapso alveolar y altera el equilibrio entre la ventilación y la perfusión, lo que ocasiona un intercambio gaseoso menos eficiente, con la consecuente hipoventilación, hipoxemia e hipercapnia.
Las alteraciones de la mecánica respiratoria no solo se limitan a los pulmones y a la pared torácica, sino que también pueden incluir la vía respiratoria superior. Se ha observado debilidad en la musculatura faríngea en la DMD, enfermedades de la neurona motora y la miastenia gravis. La debilidad del músculo dilatador de la faringe disminuye el calibre de la vía aérea superior, lo que aumenta la resistencia de esta durante la inspiración. Esto supone una mayor sobrecarga al diafragma y a los otros músculos inspiratorios, con el subsecuente aumento del trabajo respiratorio22. Durante el sueño se añade la disminución habitual del tono motor de la vía aérea superior, lo que favorece la aparición de hipoventilación y SAOS.
La pared torácicaEl patrón de restricción pulmonar que se encuentra en pacientes con ENM es a menudo mayor que el que se podría llegar a predecir, teniendo en cuenta solo la disminución de la función de la musculatura inspiratoria. La distensibilidad o compliance no es una propiedad exclusiva del pulmón, sino también de la caja torácica, y la suma de ambas conforma la distensibilidad total del aparato respiratorio. El término distensibilidad hace referencia al cambio de volumen por unidad de cambio de presión, lo que se puede apreciar gráficamente como la pendiente en una curva de presión-volumen; en otras palabras, la distensibilidad denota la facilidad con la que los pulmones y la caja torácica pueden distenderse o cambiar su forma23. El concepto opuesto sería el de la elasticidad o «retroceso elástico», que puede definirse como la tendencia para oponerse a la distensión o al cambio de forma, así como la capacidad del pulmón y la caja torácica para regresar a su volumen de reposo después de haberse distendido24.
En los pacientes con ENM se ha documentado un aumento del retroceso elástico, tanto del parénquima pulmonar como de la pared torácica25. Los mecanismos precisos que llevan a estos cambios no están bien definidos y probablemente tengan una naturaleza multifactorial. Así, la disminución crónica de la amplitud de la respiración en estos pacientes puede producir anquilosis de las articulaciones costoesternales y costovertebrales, lo que llevaría a una rigidez progresiva de la caja torácica. La fibrosis de los músculos intercostales o de elementos del tejido conectivo y las deformidades espinales también pueden reducir la distensibilidad de la caja torácica. Con la evolución del proceso, malformaciones como la escoliosis toracolumbar, que es casi universal en los niños con DMD y va empeorando con el tiempo, agravarán más aún la mecánica de la pared torácica.
Hay que señalar que en el niño pequeño con ENM lo que podemos encontrar, al contrario de lo comentado anteriormente, es un aumento de la distensibilidad, una tendencia al colapso incluso mayor que la de otros niños pequeños sin esta enfermedad, y puede que la rigidez que el pulmón va adquiriendo de forma pasiva no se alcance hasta los 4 años o más. Cuando la ENM se inicia en edades muy tempranas (AME tipo i y ii, distrofia muscular congénita, entre otras) se puede afectar la plenitud del desarrollo toracopulmonar, lo que podría influir adicionalmente en la distensibilidad pulmonar. La distorsión de la pared torácica puede ocasionar deformidades como pectus excavatum26, que pueden comprometer aún más los volúmenes corrientes. Existe la preocupación de que esta respiración a volúmenes menores desde el nacimiento pueda comprometer un desarrollo pulmonar adecuado27.
El centro de control respiratorioMuchas ENM que cursan con debilidad de la musculatura respiratoria también asocian alteraciones en la función del sistema nervioso central (SNC). La depresión del centro respiratorio puede conducir también a un fracaso de la bomba respiratoria. De hecho, el funcionamiento adecuado del SNC es particularmente importante en los pacientes con debilidad muscular, especialmente si tenemos en cuenta que el fracaso respiratorio se presenta inicialmente durante el sueño, una fase en la que se precisa un control adecuado de las respuestas del centro respiratorio. Secundariamente, además, los trastornos respiratorios del sueño pueden producir alteraciones sobre el centro de control respiratorio; en los pacientes con insuficiencia respiratoria crónica la respuesta a la hipercapnia puede ser inadecuada.
En aquellos casos en los que exista debilidad bulbar se dificulta aún más el mecanismo de la tos, comprometiendo una movilización adecuada de las secreciones. Además, se asocian alteraciones en la coordinación de la succión-deglución que pueden dar lugar a aspiraciones de repetición, infecciones respiratorias, con bronconeumonía y afectación parenquimatosa irreversible como bronquiectasias, fibrosis pulmonar21, lo que se añade a la presencia frecuente de microatelectasias en estos niños como consecuencia de la disminución de la distensibilidad pulmonar y de la caja torácica.
Una vez que la reserva respiratoria se ve comprometida, cualquier aumento de la sobrecarga puede conducir a fatiga del diafragma y fracaso respiratorio. Entre los factores que pueden aumentar esta sobrecarga se incluyen un incremento de la frecuencia respiratoria (por ejemplo, por fiebre), un aumento en la rigidez de los pulmones (por consolidación, atelectasia, etc) y/o por distensión abdominal (por ejemplo, en el contexto de una gastroenteritis).
Evaluación en consulta: clínica y exploración funcional respiratoriaEvaluación clínicaEl objetivo de la evaluación por el neumólogo del paciente con ENM es determinar el grado de extensión y progresión de la debilidad muscular y su repercusión en la función pulmonar, así como identificar comorbilidades asociadas, mediante la anamnesis, exploración física y pruebas complementarias1. Las principales complicaciones pulmonares de las ENM se expresan en la tabla 5.
Complicaciones respiratorias de las ENM
| Pérdida de función pulmonar |
| Retención de secreciones en vías aéreas |
| Alteraciones de la deglución, pérdida de la protección de la vía aérea y enfermedad pulmonar aspirativa |
| Impacto del estado nutricional |
| Impacto de la escoliosis |
| Enfermedad respiratoria durante el sueño |
| Fallo respiratorio por el día |
| Fallo respiratorio agudo |
Se deben identificar en cada visita síntomas y signos que hagan sospechar la presencia de debilidad muscular y la progresión de la misma. Hay que preguntar sobre la existencia de cambios en la fuerza de la voz o de la tos, babeo, dificultad para el aclaramiento de las secreciones, la existencia de atragantamiento con la comida o la bebida, dificultad para la masticación e intolerancia al decúbito. También se debe interrogar sobre la frecuencia y gravedad de las infecciones respiratorias, los síntomas de hipoventilación nocturna (tabla 6) y la existencia de afección respiratoria durante el sueño1.
Síntomas que deben hacer sospechar la existencia de hipoventilación
| Cefalea matutina o continua |
| Fatiga |
| Despertares con disnea o taquicardia |
| Somnolencia diurna |
| Pesadillas |
| Falta de atención |
| Síntomas y signos de fallo cardiaco derecho |
| Irritabilidad, ansiedad |
| Mal rendimiento escolar |
| Depresión |
| Dolores musculares |
| Pérdida de peso |
| Obesidad |
La exploración física debe incluir una auscultación pulmonar en la que se valorará el grado de ventilación alveolar y la existencia de secreciones retenidas, que se van a manifestar predominantemente en forma de roncus y crepitantes. La frecuencia respiratoria es un buen indicador de la función respiratoria general. Hay que tener en cuenta que estos pacientes tienen una respiración más rápida y superficial, incluso en situación estable. La valoración del estado nutricional es esencial, pues tanto la desnutrición como el sobrepeso pueden afectar de forma importante a la función respiratoria. La incoordinación toracoabdominal, sobre todo con el paciente en decúbito supino, sugiere la presencia de debilidad diafragmática.
Se debe valorar la presencia y gravedad de las alteraciones esqueléticas. La deformidad de la caja torácica de forma secundaria a la debilidad muscular ocasiona un trastorno ventilatorio restrictivo añadido al ya originado por la propia debilidad muscular. El componente de cifosis y escoliosis que se suma en estos enfermos acelera la aparición de insuficiencia respiratoria.
La valoración clínica no es capaz de predecir la existencia de alteraciones en la función pulmonar, o las alteraciones respiratorias durante el sueño, por lo que es imprescindible hacer un seguimiento reglado con pruebas específicas para estas condiciones.
Evaluación de la función pulmonarLos diversos consensos internacionales sobre el cuidado de pacientes con ENM28,29 recomiendan evaluar al menos las siguientes pruebas:
- -
Espirometría con curva flujo volumen forzada para valorar los volúmenes pulmonares y la función de las vías aéreas.
- -
Presiones inspiratorias y espiratorias máximas (PIM/PEM) y flujo pico de tos (FPT) para la valoración de la fuerza de los músculos respiratorios.
- -
Registro de pulsioximetría para detectar hipoxemia.
Estas pruebas deben comenzar a realizarse en cuanto el paciente pueda colaborar (alrededor de los 4-6 años). Se recomienda realizar los estudios de función pulmonar (espirometría, PIM/PEM y FPT) cada año hasta que se comience a detectar una alteración de las mismas. Desde este momento hasta que el paciente tenga una CVF<60% y/o PIM/PEM<60mmHg, se recomienda realizar pruebas de función pulmonar cada 6 meses y un registro domiciliario de saturación de O2 anualmente. Cuando el paciente tenga una caída de la CVF por debajo del 60% o del PIM/PEM por debajo de 60mmHg, entonces las pruebas de función pulmonar se realizarán trimestralmente, se hará un registro de saturación domiciliario cada 6 meses y una polisomnografía cada año según proponen diversos autores9,30. Pueden existir diferencias de criterios según los consensos internacionales que se analicen.
EspirometríaLa espirometría es la prueba más asequible y útil para la valoración del funcionalismo pulmonar en estos pacientes. Con un buen técnico de función pulmonar es realizable y reproducible en muchos niños por encima de 4 años. Desde un punto de vista técnico hay que prestar especial atención al sellado de la boquilla con los labios durante las maniobras espirométricas. En niños con ENM y escoliosis la medida de la talla puede no ser real o factible. En estos casos se puede utilizar la medida de la envergadura de los brazos (distancia entre las puntas de los dedos corazón con los brazos del paciente extendidos en cruz) en lugar de la talla para predecir los valores teóricos de la espirometría. En la infancia la envergadura es 1-2cm menor que la talla. A los 10 años en niños y a los 12 años en niñas se igualan talla y envergadura. Posteriormente la envergadura sobrepasa la talla en los hombres en 4cm y en las mujeres en 2cm (para una mejor estimación de la talla dividir la medida de la envergadura en niñas entre 1,01 y en niños entre 1,03).
La función pulmonar de los niños con ENM presenta típicamente un patrón restrictivo. La debilidad de los músculos inspiratorios provoca una disminución de la capacidad inspiratoria y la debilidad de los músculos espiratorios produce una disminución de la capacidad espiratoria, y la combinación de ambos produce una disminución progresiva de la CV con volumen residual normal o elevado (fig. 2). La facilidad para la medición de la CV la hace ideal para realizar medidas repetidas en la monitorización de un paciente neuromuscular. En la curva flujo-volumen existe una reducción de los flujos inspiratorios y espiratorios dependientes del esfuerzo, mostrando una curva de aspecto redondeado, con un tiempo de flujo espiratorio pico retardado y una caída brusca del flujo forzado cerca de la CRF (fig. 3). Generalmente los niños con DMD tienen un incremento de los valores absolutos de la CV con la edad hasta los 10-12 años, en que alcanzan una meseta y a partir de ahí comienza a descender. En condiciones normales la CV medida en decúbito supino no debe ser más del 10% inferior a la medida con el paciente sentado, pero cuando se produce una debilitación del diafragma esta diferencia se va incrementando. Se ha encontrado una relación clara entre la fuerza diafragmática (valorada midiendo las presiones transdiafragmáticas) y el porcentaje de caída de la CV con el paciente en decúbito supino, de forma que una CV en decúbito más de un 20% menor que sentado sugiere debilidad diafragmática significativa y el paciente tiene riesgo de sufrir hipoventilación nocturna aunque no tenga síntomas durante el día1,9.


En ENM crónicos puede existir una disminución de la CV también por reducción de la complianza pulmonar y de la caja torácica y por atelectasias pulmonares. La escoliosis va a acrecentar de forma importante la enfermedad restrictiva. Sin embargo, la estabilización de la columna no parece afectar al grado de caída progresiva de la CV en estos pacientes.
La afectación de la musculatura de la vía aérea superior puede evaluarse con la observación cuidadosa de la curva flujo-volumen. La presencia de un contorno anormal y particularmente la existencia de una meseta en el asa de flujo inspiratorio, así como oscilaciones en el flujo, puede demostrar una disfunción de la vía aérea superior31 (fig. 4).

A pesar de la reducción en los volúmenes pulmonares se observa con frecuencia un incremento del volumen residual, que probablemente refleje una alteración más intensa en los músculos espiratorios que en los inspiratorios en estos pacientes. Existe una correlación inversa entre la relación VR/CPT y la presión espiratoria máxima en boca (PEM).
Es difícil valorar si un enfermo neuromuscular puede tener además enfermedad obstructiva asociada, pues los flujos espiratorios están disminuidos más por la falta de esfuerzo muscular que por la obstrucción bronquial. Además, el incremento del VR en estos pacientes está más relacionado con la debilidad de los músculos espiratorios que con la existencia de atrapamiento aéreo debido a obstrucción bronquial. Una técnica que puede solventar este problema es la oscilometría forzada, pues al no requerir esfuerzo muscular puede valorar la existencia de una obstrucción verdadera.
La CV es un predictor de susceptibilidad a la infección: una CV<1,11l en adultos predice riesgo de infección con una sensibilidad del 90,5% y una especificidad del 70,8%32. La CV puede también predecir la supervivencia en niños y adolescentes con DMD. En niños con CV<1l la supervivencia media fue de 3,1 años y a los 5 años fue de un 8%33. Este estudio se realizó en pacientes que no recibieron tratamiento con VNI, por lo cual, actualmente, se espera una mayor supervivencia, dado que la VNI está mejorando el pronóstico de estos pacientes.
El problema que presenta la medición exclusiva de la CV es que la disminución de la misma no aparece hasta que existe una debilidad muscular pronunciada. No son necesarias presiones musculares importantes para llenar y vaciar los pulmones. Otro problema importante con la medida de la CV es que depende de una maniobra que exige una gran colaboración por parte del paciente y que cualquier enfermedad respiratoria, además de la muscular, puede producir una reducción de la CV. De todos modos la medida de la CV continúa siendo la prueba más práctica de las existentes para la valoración del niño con ENM.
Presiones máximas en bocaLa valoración de la fuerza de los músculos respiratorios es una parte importante de la evaluación funcional de un enfermo neuromuscular. La técnica más empleada es la medida de las presiones estáticas máximas en la boca: presión inspiratoria máxima (PIM) y presión espiratoria máxima (PEM). Esta técnica valora la fuerza global o la presión que los músculos respiratorios pueden generar contra una oclusión en la boca. La PIM es un índice de la fuerza diafragmática y la PEM mide la fuerza de los músculos abdominales e intercostales. El equipo requerido puede ser un manómetro o un sistema electrónico. Dado que la medida de las presiones máximas es esfuerzo dependiente, es imprescindible que el técnico sea capaz de motivar al paciente para que realice los máximos esfuerzos posibles. La PEM se mide en CPT y la PIM en volumen residual.
La técnica es sencilla. El sistema debe ser calibrado apropiadamente según las instrucciones del fabricante. Para medir la PIM se manda al paciente exhalar todo el aire hasta el volumen residual, cerrar bien los labios alrededor de la boquilla y realizar una inhalación con la máxima fuerza posible durante al menos 1s. Se deben obtener al menos 3 esfuerzos, con un máximo de 8, con los 2 más elevados que difieran menos de un 10%. Se debe permitir al paciente descansar entre 30 y 60s entre los esfuerzos. Para medir la PEM se manda al paciente realizar una inspiración máxima hasta CPT y posteriormente exhalar con la máxima fuerza posible durante al menos 1s. Se deben repetir también varias maniobras y lograr una variación menor del 10% entre las 2 mejores. Es imprescindible observar que no existan escapes de aire durante la maniobra. Los valores de ambas presiones se dan en cmH2O; la PIM es un valor negativo y la PEM positivo.
Los valores normales en niños mayores de 6 años de edad son bastante similares a los de los adultos (entre 80 y 120cmH2O) siendo algo mayor en varones. Una PIM menor de –80 o una PEM mayor de +80 excluyen la existencia de debilidad muscular respiratoria significativa. Esta medida depende completamente de la colaboración del paciente, y no siempre tienen una reproductibilidad adecuada. En un paciente bien entrenado puede ser muy útil para valorar la progresión de la enfermedad.
En los pacientes con ENM generalizada se produce una disminución similar en las presiones inspiratorias y espiratorias, sin embargo, en algunos pacientes con disminución desproporcionada de la fuerza diafragmática podemos encontrar una alteración mayor en la PIM que en la PEM. Dado que las presiones espiratorias se miden en CPT, cuando existe una disminución de la misma por la enfermedad pulmonar, puede existir una PEM exageradamente disminuida que puede hacernos subestimar la fuerza muscular. Por otro lado, cuando existe una enfermedad pulmonar obstructiva con incremento del VR, se produce una diminución de la capacidad de contracción de los músculos inspiratorios con una disminución de la PIM. Por este motivo es conveniente realizar una medición de volúmenes pulmonares mediante pletismografía (CPT), para poder evaluar correctamente los resultados, en los casos que exista enfermedad parenquimatosa pulmonar asociada a la ENM.
Flujo pico de tosLa capacidad para toser requiere el uso coordinado de músculos inspiratorios y espiratorios. La eficacia de la tos se puede evaluar determinando el FPT que se realiza con un medidor de flujo espiratorio máximo (FEM) utilizando una pieza bucal o una mascarilla facial. Se anima al paciente a realizar un esfuerzo máximo de tos (se elige el mejor de 4 a 7 intentos) y se mide el flujo máximo conseguido, reflejo de la eficacia de los músculos espiratorios. En adultos lo normal es tener un FPT superior a 350l/min. Cifras inferiores a 270l/min, tanto en situación aguda como crónica, indican deterioro en la capacidad para eliminar secreciones y establecen la necesidad de aplicar técnicas de tos asistida. En niños se ha comprobado que un FPT<160l/min predice la frecuencia de exacerbaciones pulmonares graves con una sensibilidad del 75% y una especificidad del 79%1,30.
Gases sanguíneosLa debilidad muscular progresiva provoca hipoxemia, generalmente en relación con una alteración de la relación ventilación/perfusión y posteriormente un fallo ventilatorio con hipoventilación global que provoca tanto hipoxemia como hipercapnia. Un incremento de la PaCO2 diurna por encima de 45mmHg es indicativo de hipoventilación nocturna (sensibilidad 91% pero especificidad del 75%) y un aumento del exceso de bases igual o mayor a 4mmol/l tiene una gran especificidad (100%), pero una baja sensibilidad (55%). Para evitar la realización de gasometría en la consulta podemos utilizar un pulsioxímetro y un capnógrafo, que nos darán una medida fidedigna de la SaO2 y de la PaCO2 del paciente, en condiciones basales, siempre que no exista situación de shock o anemia severa.
Trastornos del sueño en enfermos neuromuscularesLos pacientes con ENM tienen elevada predisposición a padecer trastornos respiratorios del sueño (TRS) y otros trastornos del sueño (TS), como excesiva somnolencia diurna o múltiples despertares, independientes del TRS, que proporcionan una mala calidad del sueño. Pueden presentar todo el espectro de TRS: SAOS, síndrome de resistencia aumentada de la vía aérea superior (SRAVAS), apneas centrales, caída en la saturación de O2 en sueño REM e hipoventilación en sueño REM y NREM. El patrón del TRS dependerá de la rapidez de progresión de la enfermedad y de los músculos más afectados. Diferentes mecanismos están implicados en la aparición de los TRS como debilidad de los músculos respiratorios (especialmente el diafragma) y de la vía aérea superior (VAS), patrón respiratorio restrictivo y control ventilatorio durante el sueño. Además, la mala calidad de sueño puede ser debida a dolor, disminución o ausencia de movilidad en la cama, dificultad en el aclaramiento de secreciones y trastornos asociados como ansiedad o depresión.
PrevalenciaSe estima que un niño con una ENM tiene un riesgo de TRS 10 veces superior a la población general8. Anormalidades en el intercambio de gases y disrupción de la arquitectura del sueño también son muy frecuentes. Así, en la DMD se ha encontrado que hasta un 31% presentó SAOS e hipoventilación en el 32%8. Excesiva somnolencia diurna (ESD) se ha documentado entre 33-77% de enfermos con distrofia miotónica. En la miastenia gravis se ha observado que hasta un 60% pueden padecer SAOS.
FisiopatologíaAl inicio del sueño existe una reducción en la actividad muscular de la VAS con aumento de la resistencia de la VAS. Hay un aumento de la complianza de la caja torácica y cambios en la posición y fuerza del diafragma. En sueño REM hay atonía muscular generalizada, excepto de los músculos oculares y diafragma, por tanto, el diafragma es el músculo del que dependerá la respiración. Cuando el diafragma está afectado la respiración es irregular con disminución de la ventilación minuto, menor capacidad residual funcional (CRF) y reserva de O2. Otro aspecto a recordar es que durante el sueño se produce una menor respuesta ventilatoria a la hipercapnia respecto a la vigilia, con aumento de la PaCO2 (3-4mmHg). Estos cambios fisiológicos pueden ocasionar alteraciones del intercambio gaseoso. Por todo ello, son especialmente vulnerables a TRS con múltiples factores que contribuyen a su aparición, siendo los más relevantes: debilidad del diafragma y de otros músculos, aumento de la colapsabilidad de la VAS, patrón de función pulmonar restrictivo (escoliosis, obesidad) y afectación del centro ventilatorio.
Músculos respiratorios y de la vía aérea superiorLa debilidad del diafragma condiciona hipoventilación en sueño REM y en posición supina. En vigilia utilizan músculos accesorios inspiratorios y abdominales para mantener el volumen minuto mediante elevación de la frecuencia respiratoria para compensar el volumen tidal menor. La espiración es por debajo de la CRF, permitiendo una inspiración pasiva. El mayor trabajo respiratorio conlleva a situaciones de fatiga muscular que se evidencian durante el sueño.
La colapsabilidad de la VAS puede estar incrementada por la debilidad de los músculos de la VAS, que si se asocia a factores anatómicos como anomalías craneofaciales, hipertrofia adenoamigdalar, favorece la aparición de SAOS. Se ha de pensar en SAOS en edades menores de 10 años; la adenoamigdalectomía lo mejora en muchos casos. También enfermedades con afectación predominantemente de los músculos de la VAS y afectación bulbar tienen mayor predisposición a SAOS (distrofia miotónica, neuropatías motoras-sensitivas y algunas miopatías congénitas). Además, una debilidad muscular de la VAS muy acentuada con preservación del diafragma condiciona que la colapsabilidad se incremente al generar el diafragma una elevada presión negativa inspiratoria.
Un patrón restrictivo pulmonar es frecuente en ENM asociado a debilidad muscular, escoliosis y obesidad. La escoliosis es una complicación muy frecuente que reduce la complianza de la caja torácica, favorece la presencia de hipoventilación y atelectasias, altera la relación ventilación/perfusión pulmonar y aumenta el trabajo respiratorio. La estabilización de la columna vertebral reduce la morbilidad respiratoria al preservar la mecánica pulmonar. Por otra parte, la obesidad es un riesgo conocido para presentar SAOS (mayor colapsabilidad de la VAS, menor volumen pulmonar…).
Centro respiratorioEn general, los sujetos con ENM tienen una respuesta ventilatoria normal34. No obstante, en algunas miopatías congénitas puede existir un trastorno del control ventilatorio per se. Por otra parte, una hipoventilación crónica conduce a fatiga muscular y tolerancia de la hipercapnia debido a menor respuesta ventilatoria por alteración de los quimioreceptores. La alteración de la respuesta ventilatoria a los niveles de CO2 por un lado beneficia al elevar el umbral de arousal, y evitar así la disrupción del sueño, pero por otra parte prolonga los eventos con hipoventilación pronunciada.
Alteración de mecanismos regulatorios del sueñoUna excesiva somnolencia diurna es común en los pacientes con ENM y esta puede presentarse sin SAOS asociado. Así, la distrofia miotónica puede presentar somnolencia severa e inicio del sueño en REM, rasgos típicos de la narcolepsia; algunos presentan niveles bajos de hipocretina, con lo que se ha postulado una afectación hipotalámica.
Tipos de trastornos respiratorios del sueño- -
SAOS. Los eventos obstructivos son más frecuentes en sueño REM, y cuando hay afectación mayor de músculos respiratorios accesorios y de VAS. Suele preceder a la hipoventilación.
- -
Hipoventilación. Es característico que al inicio solo se produzca en sueño REM; al avanzar la enfermedad afecta a todas las fases del sueño. Puede estar presente a pesar de normocapnia diurna. Es habitual cuando se afecta el diafragma. Si se eleva el umbral de arousal, como consecuencia de una hipoventilación crónica no tratada, se permite largos y pronunciados períodos de hipoventilación sin despertares que conducen a fallo ventilatorio e hipertensión pulmonar y sobrecarga cardíaca.
- -
Sueño fragmentado. El umbral de arousal condiciona que se produzca un despertar ante elevaciones de CO2, y también ante presión intratorácica negativa en un intento de abrir la VAS. Aunque el arousal es un mecanismo protector, un elevado número de despertares reducen el tiempo total de sueño (TTS), así como el sueño REM y el sueño de ondas lentas (reparador). Se manifiesta con somnolencia y fatiga diurna.
La distribución de la debilidad muscular y la rapidez de progresión de la ENM influyen en el tipo de TRS. Generalmente, los TRS se correlacionan con la severidad de la debilidad muscular. Por tanto, el tipo y grado de músculos afectos, además de la edad del paciente, son factores determinantes en la gravedad del TRS.
Distrofia muscular de DuchenneEn fases precoces hay afectación de músculos de la VAS e intercostales, con preservación del diafragma que progresivamente se afecta también. Así, hasta los 10 años de edad, es más frecuente el SAOS (habitualmente se resuelve con adenoamigdalectomía), y posteriormente la hipoventilación nocturna.
Atrofia muscular espinalSuele estar preservado el diafragma, y hay mayor afectación de los músculos intercostales, pudiendo observarse desaturación en sueño REM sin hipercapnia con anterioridad a la hipoventilación de fases más avanzadas.
Miastenia gravisLos TRS son muy frecuentes; predomina el SAOS.
Distrofia miotónicaSon muy frecuentes los TRS, pudiendo presentar todo el espectro. Además, hasta un 33% de los niños presentan movimientos periódicos de piernas que producen sueño fragmentado.
Evaluación clínicaLa historia clínica debe investigar la presencia de ronquido, sueño inquieto, arousals frecuentes, sudoración nocturna, somnolencia diurna, cefalea y letargia matutina. Se debe valorar también las consecuencias de los TRS como estancamiento ponderal y trastornos neurocognitivos. Estos síntomas son inespecíficos, de inicio insidioso e incluso se consideran erróneamente como parte de la propia evolución de la enfermedad. Además, síntomas como fatiga, problemas de deglución, tos ineficaz, pérdida de peso e infecciones respiratorias frecuentes sugieren empeoramiento del trastorno muscular con agravamiento de los posibles TRS presentes. Los cuestionarios clínicos y de calidad de vida son útiles para caracterizar y monitorizar de forma sistemática los síntomas y consecuencias de los TRS, además de evaluar la respuesta al tratamiento con VNI.
Se utilizan como criterios predictores de hipoventilación durante el sueño: FVC<60%, PaCO2>45mmHg y EB>4mmol/l diurnos35. La caída de la CV en supino es indicativa de afectación diafragmática. Sin embargo, la espirometría es poco sensible y estos valores de intercambio gaseoso son sugestivos de hipoventilación ya instaurada. Parece más apropiado valorar la presencia de TRS con una prueba que evalúe la ventilación durante el sueño. La polisomnografía (PSG) es la prueba más precisa y fiable para el diagnóstico de los TRS. Si no se dispone de PSG una pulsioximetría nocturna junto con una monitorización continua de CO2 (capnografía) es una alternativa válida. Es aconsejable también titular los parámetros ventilatorios del paciente en el laboratorio de sueño si es posible y monitorizar la ventilación periódicamente, ya que los requerimientos ventilatorios pueden cambiar a lo largo de su evolución y en determinadas situaciones.
Manejo de los trastornos del sueñoNo está establecido claramente cuándo comenzar la VNI. Se considera apropiado iniciar VNI ante la presencia de SAOS (en caso de que no se resuelva con cirugía adenoamigdalar) y/o hipoventilación, objetivado por estudio de sueño. Se define hipoventilación nocturna en niños si PaCO2>50mmHg durante>25% del TTS (medida por capnografía transcutánea o espirada)36. En adultos también definen hipoxemia durante el sueño si la saturación nocturna de O2 es<90% durante >5min con nadir de 85% o al menos>30% del TTS con saturación nocturna de O2<90%. Si se detecta hipoxemia no se aconseja administrar oxígeno sin ventilación complementaria, ya que la hipoxemia suele asociarse a hipoventilacion, y se agravaría la hipercapnia. Una indicación evidente de inicio de VNI es PaCO2>45mmHg diurna, aunque es preferible actuar antes y no obviar la hipoventilación nocturna precedente. La VNI estabiliza la VAS, mejora la eficiencia del sueño, la mecánica pulmonar, normaliza gases arteriales y reduce el trabajo respiratorio. Los TRS preceden al fallo ventilatorio diurno; estudios recientes apuntan a que el diagnóstico y tratamiento precoz de estos contribuyen a mejorar el pronóstico de estas enfermedades y mejorar su calidad de vida.
La presión continua positiva en la vía aérea (CPAP) debe usarse cuando existe SAOS, y en la hipoventilación se recomienda ventilación a 2 niveles de presión (BIPAP). Ante una ESD, corregido ya el TRS, se puede dar modafinilo, por ejemplo en la distrofia miotónica. Otros aspectos a tratar son el adecuado control del dolor y rigidez muscular, posición y movilidad en la cama.
Otras comorbilidades en las enfermedades neuromuscularesCardiopatíaMuchas de las ENM pueden tener repercusión cardiológica, en ocasiones con gran relevancia clínica. La miocardiopatía dilatada o hipertrófica, así como los trastornos del ritmo y de la conducción son las principales manifestaciones, que varían en función de la enfermedad de base37.
Distrofia muscular de DuchenneLa afectación cardíaca en esta enfermedad es frecuente y precoz, pudiendo aparecer las primeras manifestaciones alrededor de los 6 años de edad, y estando presentes al final de la adolescencia entre el 95-100% de los casos.
Es habitual encontrar alteraciones del ritmo y de la conducción, siendo la taquicardia sinusal inapropiada la manifestación más frecuente. La presencia de ondas R altas o cociente R/S incrementado en precordiales derechas, ondas Q anormales y ondas T melladas es habitual en el electrocardiograma (ECG). Entre los trastornos de conducción los defectos de conducción intraauricular e infranodales son los más comunes. En la ecocardiografía es posible detectar los hallazgos típicos de una miocardiopatía dilatada, como dilatación ventricular derecha e izquierda y disminución de la fracción de eyección. La alteración ecocardiográfica más precoz es la alteración de la distensibilidad del ventrículo izquierdo.
Las alteraciones en el ECG y la ecocardiografía acostumbran a ser subclínicas hasta los 10 años de edad, y habitualmente no progresan o lo hacen lentamente. La intensidad de la miocardiopatía no está en relación con la severidad del resto de enfermedad. La evolución de la enfermedad en comparación con la lenta progresión de la afectación cardíaca comporta que únicamente la taquicardia sinusal inapropiada acostumbre a poder tener relevancia clínica. En un 40% de los casos en estadios avanzados de pacientes no ventilados aparece fallo cardíaco, que parece más relacionado con el fracaso ventilatorio y la hipertensión pulmonar que con la propia cardiopatía.
Enfermedad de BeckerEntre un 65-75% de los pacientes afectados por la enfermedad de Becker presentan alteraciones a nivel cardíaco, pudiendo producirse en estadios preclínicos de la misma. Los hallazgos característicos en el ECG de estos pacientes son los mismos que en los afectados por la DMD. En la ecocardiografía pueden observarse signos de miocardiopatía dilatada, también habituales en los pacientes con DMD.
La incidencia de la miocardiopatía se incrementa con la edad, aunque la gravedad es independiente a ella. El grado de afectación a nivel cardiológico tampoco tiene relación con la intensidad de los síntomas extracardíacos. A diferencia de la DMD, la miocardiopatía puede comprometer la vida del paciente, puesto que las manifestaciones extracardíacas son menos relevantes y tienen menos repercusión en la esperanza de vida de los pacientes.
Atrofias musculares espinalesPrincipalmente se han descrito alteraciones cardiológicas en la AME tipo iii. Las principales manifestaciones son la miocardiopatía dilatada, alteraciones en la conducción (especialmente bloqueo AV completo), fibrilación auricular y parálisis auricular. También pueden observarse signos electrocardiográficos de sobrecarga del ventrículo derecho por hipertensión pulmonar secundaria a los trastornos ventilatorios de la enfermedad.
Trastornos nutricionales y de la degluciónLas alteraciones a nivel digestivo en los ENM son muy frecuentes, y en muchas ocasiones infravaloradas. En una revisión de 451 pacientes de edad pediátrica y adultos afectados por diversas ENM (DMD, distrofia muscular facio-escápulo-humeral, AME, miastenia gravis, dermatomiositis y polimiositis), el 34,9% de ellos padecían algún tipo de trastorno de la alimentación38. De forma global, las alteraciones que con mayor frecuencia presentan estos pacientes son limitaciones en la apertura de la boca, alteraciones de la masticación, de la deglución y reflujo gastroesofágico (RGE). Si nos centramos en pacientes afectados por la DMD, aproximadamente el 10% de ellos sufre problemas en la apertura de la boca y un 20% dificultades en la masticación39. En pacientes con AME tipo ii los porcentajes que se hallaron de pacientes afectados por estas alteraciones son similares40. En ambos casos la frecuencia de aparición de estas complicaciones aumenta con la edad. Estas afectaciones pueden tener como consecuencia trastornos de la deglución que pueden comportar, entre otras complicaciones, alteraciones nutricionales y sobreinfecciones respiratorias secundarias a aspiraciones. Es por este motivo que es de gran importancia el abordaje multidisciplinar de estas enfermedades, con una evaluación seriada de la ingesta de los pacientes, una exploración física completa con parámetros antropométricos y análisis con marcadores nutricionales.
Una deglución normal se divide en 3 fases: oral, faríngea y esofágica. Durante la fase oral la boca, la lengua y los dientes forman el bolo alimenticio que posteriormente se impulsa hacia atrás mediante la lengua. El cierre de la laringe confiere protección a las vías respiratorias. La laringe realiza un movimiento hacia arriba y adelante que, además de proporcionar una protección adicional a las vías respiratorias, mantiene relajado el esfínter esofágico superior. La disminución de presión de este esfínter posibilita el desplazamiento del bolo desde la base de la lengua hasta faringe inferior y esófago superior. El peristaltismo iniciado en la base de la lengua y la pared faríngea posterior despeja la faringe de los residuos del bolo.
La disfagia en enfermedades musculares crónicas se debe principalmente a la debilidad muscular41. La debilidad en la lengua, musculatura de la cara y mandíbula pueden deteriorar la capacidad de preparar el bolo adecuadamente y recuperar las partículas del mismo. La debilidad palatina puede favorecer la regurgitación nasal. La debilidad de los músculos suprahioideos ocasiona un deterioro de la apertura del esfínter esofágico superior. Esto, a su vez, produce un deterioro en el tránsito del bolo, acumulación en la faringe y un mayor riesgo de aspiración en la laringe a consecuencia de la imposibilidad de un cierre adecuado y la falta de una tos eficaz. Cualquier debilidad muscular respiratoria compromete los mecanismos de defensa de las vías respiratorias y deteriora el reflejo de la tos, facilitando las complicaciones respiratorias.
El hecho de que el origen de la disfagia se encuentre en la debilidad muscular conlleva que estos trastornos de la deglución impliquen con más frecuencia los alimentos sólidos que los semisólidos o líquidos. Si nos centramos en una revisión realizada en pacientes afectos de DMD, de los 118 pacientes 5 presentaban atragantamiento con sólidos, semisólidos y líquidos, 12 únicamente con sólidos y 4 con líquidos39. La aparición de estos eventos aumentaba con la edad de los pacientes, presentando clínica con todas las texturas únicamente un paciente menor de 18 años, 4 únicamente con sólidos y ninguno con semisólidos o líquidos.
La evolución del peso de los pacientes con ENM varía en función de la enfermedad de base y del estadio de la misma. Estudios realizados en la DMD han observado que los pacientes mantienen el peso adecuado para su edad en referencia a la población sana durante la primera década de la vida. En una segunda fase, situada alrededor de los 13 años y coincidiendo con la pérdida de capacidad para la deambulación, los pacientes tienden a aumentar su peso. El aumento de peso produce un impacto negativo en la progresiva pérdida de función motora y en el desarrollo de escoliosis. Finalmente, la evolución de los pacientes mayores tiende a ser a la pérdida de peso, observándose una tendencia a presentar un peso por debajo de 2 DS en relación con la población sana del mismo grupo de edad a partir de los 18 años. La progresiva pérdida de peso es achacable al aumento de las dificultades en la alimentación, pero también al empeoramiento del estado respiratorio, tanto por lo que hace referencia al aumento de episodios de sobreinfección respiratoria como a la presencia de hipoventilación nocturna, que se ha demostrado como causa que impide una normal ganancia de peso. La utilización de VNI ante los primeros signos sugestivos de fallo respiratorio ha demostrado favorecer que estos pacientes se mantengan con un peso adecuado.
La evaluación clínica es necesaria para tomar decisiones sobre las mejores medidas en el manejo de la alimentación de estos pacientes, pero puede ser insuficiente para valorar la fase oral y faríngea de la deglución. La videofluoroscopia permite la evaluación de estas fases con las distintas consistencias de los alimentos. Mediante esta exploración es posible observar la formación y manipulación del bolo en la fase oral y su desplazamiento hacia la faringe para la deglución. Posteriormente nos posibilita la detección de residuos en la faringe, conociendo su volumen y localización exacta. Finalmente nos proporciona información sobre la penetración de material alimentario hacia las vías respiratorias42.
La fibroendoscopia de la deglución (FEES) consiste en realizar una observación directa de la deglución situando el fibrobroncoscopio en la hipofaringe y ofreciendo al paciente las diferentes texturas alimentarias teñidas con colorante. Ofrece información sobre la anatomía laríngea, manejo de las secreciones salivares, movilidad de las cuerdas vocales, permite comprobar la sensibilidad laríngea mediante pulsos de aire y finalmente la aspiración. Con respecto a la videofluoroscopia ofrece las ventajas de la información anatómica descrita, la realización a la cabecera del paciente y que además al no irradiar permite valorar la respuesta a los consejos terapéuticos ofrecidos, como por ejemplo cambios de textura o postura. Como inconvenientes tiene que no valora la fase oral o esofágica. La concordancia intraoperador e interoperador es similar a la videofluoroscopia.
En la literatura no existe un consenso sobre cuál es el gold standard en el diagnóstico de aspiración, por lo que su uso dependerá de la disponibilidad de cada centro, siempre teniendo en cuenta las limitaciones de cada una y que en caso necesario pueden ser incluso complementarias.
En los casos en los que ni la videofluoroscopia y la FEES permitan el diagnóstico de aspiración puede ser de utilidad el recuento de macrófagos alveolares cargados de lípidos en un lavado broncoalveolar. El diagnóstico se obtiene mediante un índice consistente en asignar un valor entre 0 y 4 a cada macrófago, en función de la cantidad de lípido en su citoplasma (tabla 7). Se hace el recuento sumando los valores de 100 macrófagos, siendo sugestivo de aspiración un valor superior a 100. Nuevos estudios sugieren que elevando el valor de corte a 150 se mejora la especificidad sin apenas afectar la sensibilidad43.
El abordaje terapéutico de estos pacientes requiere un equipo multidisciplinar bien coordinado con la presencia de neumólogos, gastroenterólogos, logopedas y fisioterapeutas. Una revisión de la Cochrane41 muestra que no existen estudios aleatorizados que permitan llegar a una conclusión sobre la mejor estrategia terapéutica. A pesar de esto, parece claro que el primer paso en el tratamiento de estos pacientes debe consistir en cambios posturales durante la ingesta, cambios en el tamaño del bolo alimenticio y en la consistencia de los alimentos ingeridos. Tal y como se ha comentado previamente, los pacientes afectos de ENM acostumbran a presentar más problemas con los alimentos sólidos, por lo que es recomendable, ante la presencia de alteraciones de la deglución, favorecer los alimentos de consistencia líquida y pastosa. En los casos en los que estas medidas no sean suficientes y persistan problemas en la deglución y múltiples procesos infecciosos por aspiración, es recomendable limitar o eliminar la ingesta por vía oral. Las principales formas de alimentación por vía enteral son la sonda nasogástrica y la gastrostomía percutánea endoscópica, siendo mucho más utilizada y mejor tolerada por parte de los pacientes esta segunda opción. La utilización de la gastrostomía permite, además de reducir los episodios de aspiración y por consiguiente de infecciones respiratorias, un aumento de peso de los pacientes. Un estudio observacional de niños con distrofia muscular alimentados mediante gastrostomía aumentaron su peso del percentil 3 al percentil 10. Es importante también la administración de suplementos nutricionales desde las primeras fases de la enfermedad a los pacientes con pérdida de peso u otros déficits nutricionales.
Enfermedad por reflujo gastroesofágicoEl RGE es, junto con las alteraciones descritas previamente, una manifestación digestiva habitual en los pacientes con ENM. En condiciones normales el esfínter esofágico inferior es la principal barrera para evitar el paso de contenido gástrico hacia el esófago. Este esfínter es un círculo de musculatura lisa en el extremo distal del esófago que permanece cerrado en situación de reposo y se relaja para permitir su apertura en la deglución o con el estímulo de la distensión del fundus gástrico44. La debilidad muscular habitual en estos pacientes conduce con frecuencia a una menor competencia del esfínter esofágico inferior. El principal mecanismo que comporta la aparición de reflujo en estos pacientes es la presencia de una gastroparesia. Esto comporta un vaciamiento del estómago retardado, y por consiguiente una distensión gástrica que se traduce en la aparición de reflujo. Se ha observado que una de las principales complicaciones de los pacientes portadores de gastrostomía es el RGE causado por la distensión gástrica.
Un cierto grado de reflujo es fisiológico en todas las personas. Hablamos de enfermedad por reflujo gastroesofágico (ERGE) cuando este reflujo comporta manifestaciones clínicas o una inflamación de la mucosa esofágica. La clínica puede ser digestiva, especialmente pirosis, pero también respiratoria en forma de infecciones de repetición, broncoespasmo o tos crónica. En los pacientes con ENM son especialmente importantes estas últimas manifestaciones por el déficit de sistemas de protección de las vías respiratorias descritos anteriormente.
El uso del tránsito gastroesofágico para el diagnóstico del reflujo es limitado, dada la poca sensibilidad de esta exploración. La principal herramienta diagnóstica de esta enfermedad es la medición del pH intraesofágico mediante la pHmetría.
El abordaje terapéutico de esta enfermedad puede ser inicialmente farmacológico. Los fármacos más comúnmente utilizados son los inhibidores de la bomba de protones, como el omeprazol y los procinéticos, como la domperidona. Es frecuente que el control de la enfermedad mediante los fármacos sea insuficiente, siendo necesario recurrir a la cirugía. La técnica quirúrgica de elección es la funduplicatura de Nissen. La alta incidencia de RGE en pacientes con gastrostomía hace que sea frecuente la realización de esta técnica quirúrgica en pacientes que reciben alimentación por esta vía. Cabe destacar que la recurrencia del reflujo en los pacientes con ENM intervenidos quirúrgicamente es muy superior al resto de la población45.
ConclusionesPara mejorar la supervivencia y la calidad de vida de los pacientes con ENM debemos proponernos los siguientes objetivos: prevenir las infecciones respiratorias, vigilar el estado nutritivo, prevenir las deformidades torácicas, tratar las comorbilidades (SAHOS, RGE, IRA, etc.), extremar los cuidados perioperatorios (cirugía de escoliosis, destete del ventilador) y vigilar la instauración de la IRC. A este primer artículo, en el que se ha descrito la fisiopatología respiratoria de la ENM y de sus comorbilidades, le sigue un segundo artículo sobre los tratamientos respiratorios que precisan estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los miembros del Grupo de Técnicas de la Sociedad Española de Neumología Pediátrica (SENP): D. Álvarez, V. Alzina, A. Andrés, O. Asensio, I. Barrio, J. Blanco, A. Bonillo, M. Bosque, E. Bragado, G. Cabrera, P. Caro, M. Carrasco, R. Cilveti, M. Cols, J. Corominas, I. Cortell, I. de Mir, O. de la Serna, Y. Delgado, C. Díaz, J. Elorz, A. Escribano, J. Figuerola, M. Gaboli, D. Gómez-Pastrana, A. López, M. Machuca, A. Moreno, L. Moreno, C. Oliva, L. Pardos, T. Pascual, D. Pastor, A. Peñas, E. Pérez, G. Pérez, J. Pérez, C. Reverte, S. Rovira, A. Salcedo, E. Sánchez, J. Sánchez, L. Sanz, V. Sanz, O. Sardón, J. Sirvent, J. Tabares, A. Torrent, J. Torres e I. Úbeda.
