






Introducción
El déficit de expresión de moléculas de clase II del complejo mayor de histocompatibilidad (MHC-II) es una inmunodeficiencia primaria combinada. Su herencia es tipo autosómico recesivo. Es poco frecuente, siendo la mayoría de los casos procedentes de la zona norteafricana. Existe una ausencia de moléculas MHC-II en la superficie de las células que habitualmente las expresan, confiriendo una inmunodeficiencia grave celular y humoral, con susceptibilidad extrema a infecciones bacterianas, virales, fúngicas y protozoarias1,2. Su diagnóstico se basa en la sospecha clínica y el estudio inmunitario. El tratamiento se basa fundamentalmente en la profilaxis de infecciones oportunistas a la espera de un trasplante de progenitores hematopoyéticos (TPHA), siempre que se encuentre un donante compatible1. Si no se realiza un diagnóstico y tratamiento precoz, esta patología suele resultar letal durante los primeros 2 años de vida debido a infecciones y fallo de medro graves.
Material y métodos
Se revisaron las historias clínicas de los 4 pacientes diagnosticados de déficit de MHC-II en la Unidad de Infectología e Inmunodeficiencias Pediátricas de nuestro hospital entre los años 1998 y 2005. Los criterios diagnósticos son los propuestos la Sociedad Europea de Inmunodeficiencias (European Society for Immunodeficiencies, ESID)3 (tabla 1). En cada caso se han recogido las características epidemiológicas, manifestaciones clínicas, pruebas complementarias realizadaspara su diagnóstico, tratamientos recibidos y evolución clínica hasta el momento actual.

Observación clínica
Se diagnosticaron 4 casos cuyas características epidemiológicas se describen en la tabla 2. Tres de los casos procedían de Marruecos. En 2 casos existía consanguinidad, todos ellos con antecedentes familiares de fallecimientos por causa infecciosa en el primer año de vida.

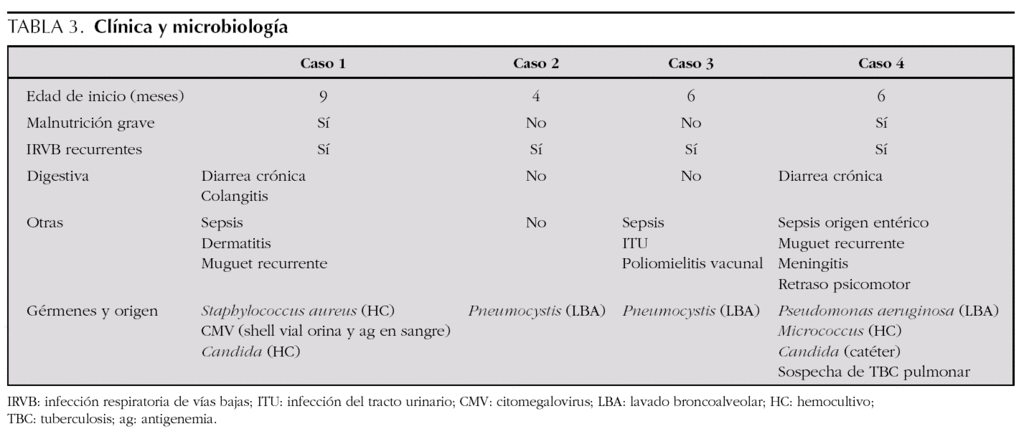
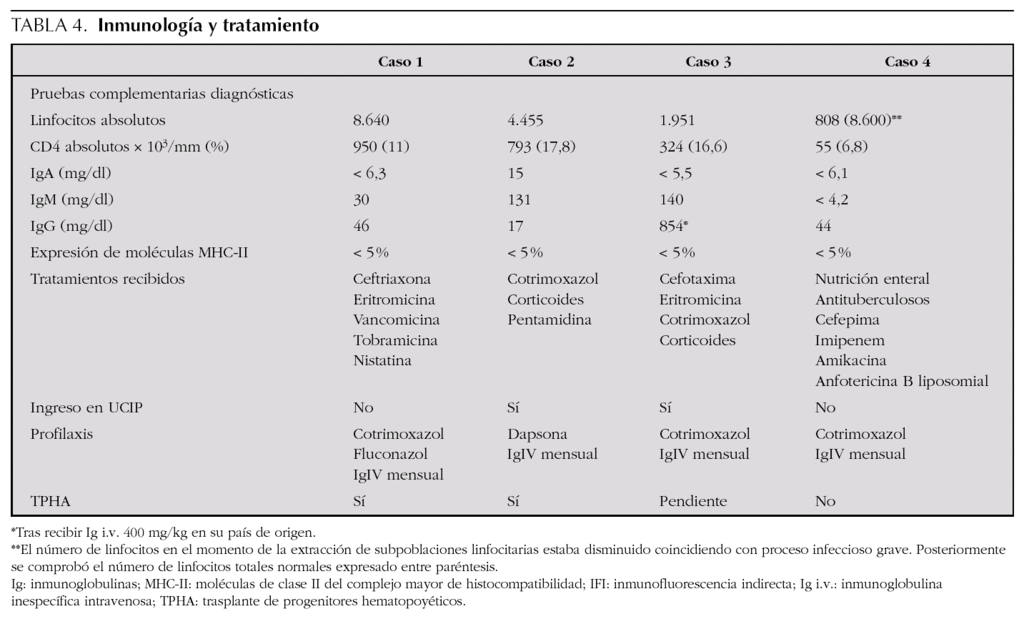
En cuanto a la clínica (tabla 3), todos los casos empezaron con infecciones durante el primer año de vida. Las manifestaciones respiratorias se presentaron en todos los pacientes, y las manifestaciones digestivas, como la diarrea crónica o la colangitis, así como la malnutrición, estaban presentes en la mitad. Otras manifestaciones clínicas fueron las sepsis bacterianas, presentes en tres de ellos, y las infecciones del sistema nervioso central que aparecieron en dos. Los gérmenes causantes de los cuadros clínicos incluyen bacterias como Escherichia coli o Staphylococcus aureus, hongos y gérmenes oportunistas (tabla 3).Las pruebas complementarias diagnósticas, destacadas en la tabla 4, aportan en todos los casos unas subpoblaciones linfocitarias caracterizadas por una clara disminución de los linfocitos CD4+, aunque con un número total de linfocitos conservados, acompañado en la mayor parte de los casos de hipogammaglobulinemia.Los tratamientos recibidos (tabla 4) incluyeron en todos los casos antibioterapia combinada según los gérmenes implicados y antibiograma, al ser la patología infecciosa la más frecuente. También requirieron soporte nutricional y respiratorio algunos pacientes, precisando 2 casos ingreso en la unidad de cuidados intensivos pediátricos para ventilación mecánica por neumonía grave por Pneumocystis jirovecii. Tras el diagnóstico de la inmunodeficiencia, todos los pacientes recibieron quimioprofilaxis con inmunoglobulina intravenosa inespecífica sustitutiva (400 mg/kg) cada 21-28 días y cotrimoxazol diario o a días alternos (sustituido por dapsona en uno de ellos por presentar una reacción alérgica).En dos de los casos se consiguió realizar TPHA-HLA idéntico procedente de alguno de los hermanos, presentando buena evolución tras 4 y 6 años de seguimiento respectivamente. En el caso 3 se perdió el seguimiento tras el diagnóstico, aunque posteriormente supimos que había fallecido por causa infecciosa en otro centro hospitalario y el caso 4 falleció como consecuencia de una candidiasis invasiva refractaria a diferentes terapias antifúngicas.
Discusión
El déficit de MHC-II es una inmunodeficiencia combinada poco frecuente y de herencia autosómica recesiva. Los primeros pacientes afectados de este cuadro se comunicaron a finales de los años 1970 y principios de 19804-6. En el registro español de inmunodeficiencias primarias (REDIP)7, figuran escasísimos casos aportados. En las familias afectadas hay una alta incidencia de consanguinidad y una gran parte de los afectados proceden del norte de África2, como en los casos que describimos.
La enfermedad se caracteriza por una falta de expresión de MHC-II en las células presentadoras de antígenos, provocando un cuadro de inmunodeficiencia combinada grave (celular y humoral). Los genes afectados no son exactamente los que codifican a las moléculas del MHC-II, sino aquéllos relacionados con las proteínas que promueven la transcripción de dichas moléculas8. La alteración final puede ser el producto de la mutación de 4 grupos de genes diferentes, dando lugar a los 4 grupos actualmente descritos para esta enfermedad, que se denominan grupo A (gen CIITA), B (gen RFX5), C (gen RFXAP) o D (gen RFXANK)9,10. Esto explica que exista cierta heterogeneidad en cuanto a la gravedad y momento de aparición de las manifestaciones clínicas. Incluso se ha descrito un fenotipo inusual en el cual existe una expresión residual de HLA DRα y β asociado con un número normal de linfocitos CD4 con un mejor pronóstico11,12.
Las infecciones recurrentes incluyen tanto infecciones bacterianas, sobre todo bacilos gramnegativos (Pseudomonas, Salmonella, E. coli, Proteus), como virales, fúngicas (Candida albicans, Pneumocystis jirovecii) o protozoarias (Cryptosporidium parvum, Giardia lamblia)1.
El cuadro clínico suele comenzar en los primeros 6-12 meses de vida, predominando la sintomatología infecciosa gastrointestinal y respiratoria, provocando diarrea crónica, malabsorción y fallo de medro1. Es característica la aparición de hepatitis y colangitis esclerosante en estos pacientes, esta última asociada normalmente a infección intestinal crónica por Cryptosporidium.
En ocasiones, se producen manifestaciones neurológicas, debidas en su mayor parte a agentes virales como Coxsackie, adenovirus o poliovirus. Este último suele ser de origen vacunal, como el caso que aportamos. Hay que recordar por tanto esta posibilidad en este tipo de pacientes si proceden de países africanos, dado que allí se sigue vacunando con polio oral. En algunas ocasiones se producen cuadros de meningoencefalitis13, que suelen ser fulminantes o dejar secuelas.
La progresión suele ser rápida, llevando sin tratamiento al fallecimiento en los primeros 5 años de vida, aunque casi siempre dentro de los dos primeros10,13-15. En muchas ocasiones se observan casos de sepsis, llevando a la muerte precoz sobre todo en zonas menos favorecidas como el norte de África.
Su diagnóstico se basa en la sospecha clínica8 ante un lactante con clínica compatible con o sin antecedentes familiares y procedente de áreas de mayor prevalencia. Los datos de laboratorio y estudios inmunológicos en estos pacientes mostrarán10,13: a) número de linfocitos T y B totales normal pero con un número de linfocitos T CD4+ reducido, con cribado negativo para infección por virus de la inmunodeficiencia humana (VIH); b) hipogammaglobulinemia con disminución de uno o dos isotipos, al estar la respuesta humoral reducida o ausente, y c) expresión reducida (< 5 %) de las moléculas MCH-II en células B y monocitos, prueba habitualmente al alcance de todos los departamentos de inmunología de nuestro medio. El diagnóstico definitivo será posible tras detectar la mutación de algunos de los genes implicados y citados anteriormente.
Desde el punto de vista terapéutico, es una indicación absoluta de TPHA como única alternativa curativa1,8, obteniéndose buenos resultados con el trasplante procedente de familiar HLA idéntico, sobre todo cuando se realiza antes de los 2 años de vida1. El éxito del mismo puede ser obstaculizado fundamentalmente por las infecciones virales persistentes, el rechazo del injerto o la enfermedad injerto contra el huésped. En espera del trasplante, es fundamental el uso de antibióticos profilácticos, inmunoglobulinas intravenosas y un aporte nutricional adecuado13,16. Quizás en un futuro la terapia génica pudiera representar un alternativa aún mejor8.
Correspondencia: Dra. M.ªM. Serrano-Martín.
Unidad Infectología e Inmunodeficiencias. Departamento de Pediatría.
Hospital Materno-Infantil Carlos Haya.
Avda. Arroyo de los Ángeles, s/n. 29011 Málaga. España.
Correo electrónico: mmarser@hotmail.com
Recibido en enero de 2006.
Aceptado para su publicación en septiembre de 2006.
