




Sr. Editor:
La colestasis intrahepática familiar progresiva tipo 3, o defecto de MDR3, es un trastorno colestásico hereditario crónico autosómico recesivo, aunque se han descrito mutaciones de novo, que se inicia en los primeros meses de vida y puede evolucionar a cirrosis hepática en la primera década.
Informamos del caso de un niño de 2 años y 8 meses de edad, que desde los 12 meses presentaba ictericia, coluria e hipocolia sin prurito. Es el sexto hijo de padres no consanguíneos fruto de un embarazo bien controlado que cursó sin ictericia ni prurito. El parto fue a término, eutócico, pesó al nacer 3 000g y el período neonatal transcurrió sin incidencias. Sus 5 hermanos están sanos.
A su ingreso presentaba buen estado general, bien nutrido, ictericia cutaneomucosa, abdomen distendido, hepatomegalia de 3–4cm, de consistencia dura y esplenomegalia de 6–7cm, con red venosa superficial visible. No se observaban rasgos dismórficos ni soplos. En la analítica destacaba hipertransaminasemia (GOT 211 U/l, GPT 156 U/l, GGT 789 U/l), elevación de los ácidos biliares (450μmol/l, VN [0–6]) e hiperbilirrubinemia a expensas de bilirrubina directa (9,2mg/dl); el estudio de autoinmunidad, serología de virus hepatotropos, inmunoglobulinas y α1-antitripsina fueron normales. En la ecografía se evidenciaban datos de hipertensión portal, flujo portal hepatópeto y esplenomegalia homogénea de 10,5cm. Los estudios histológico e inmunohistoquímico mostraron proliferación ductal, cúmulo de pigmento biliar en el citoplasma de hepatocitos y células de Kupffer y minúsculos nódulos hepatocitarios, carentes de espacios porta, incluidos en una densa trama fibrosa que conecta entre sí los espacios porta junto con ausencia de tinción canalicular normal para MDR3-P gp (fig. 1). La evolución de nuestro paciente, pese al tratamiento con ácido ursodesoxicólico (UDCA) y soporte nutricional, ha sido desfavorable, y actualmente está incluido en la lista de espera para trasplante hepático.
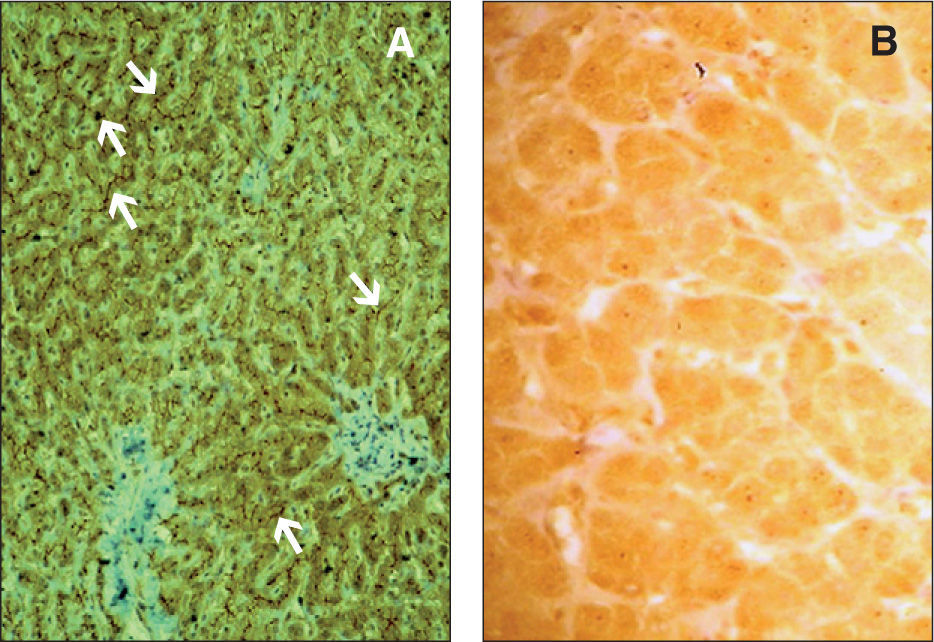
Tinción inmunohistoquímica para MDR3-P gp en tejido hepático. A) Tinción canalicular normal (flechas). B) Ausencia total de tinción canalicular, defecto de MDR3 (por cortesía de los doctores P. Jara, del Servicio de Hepatología y Trasplante Hepático Infantil, y L. Álvarez, de la Unidad de Investigación, ambos del Hospital Universitario La Paz. Madrid).
El ABCB4, gen responsable del defecto de MDR3, está localizado en el cromosoma 7 (7q21), y codifica una P-glucoproteína de membrana denominada proteína de resistencia a multifármacos 3 (MDR3), que en condiciones normales se expresa predominantemente en la membrana canalicular del hepatocito y, con menor intensidad, en la glándula suprarrenal, el corazón, el músculo estriado, el bazo y las amígdalas1. La proteína MDR3 interviene en la excreción canalicular de la fosfatidilcolina, cuya función es la formación de micelas mixtas e impedir el daño canalicular secundario a la acción detergente de las sales biliares (fig. 2).
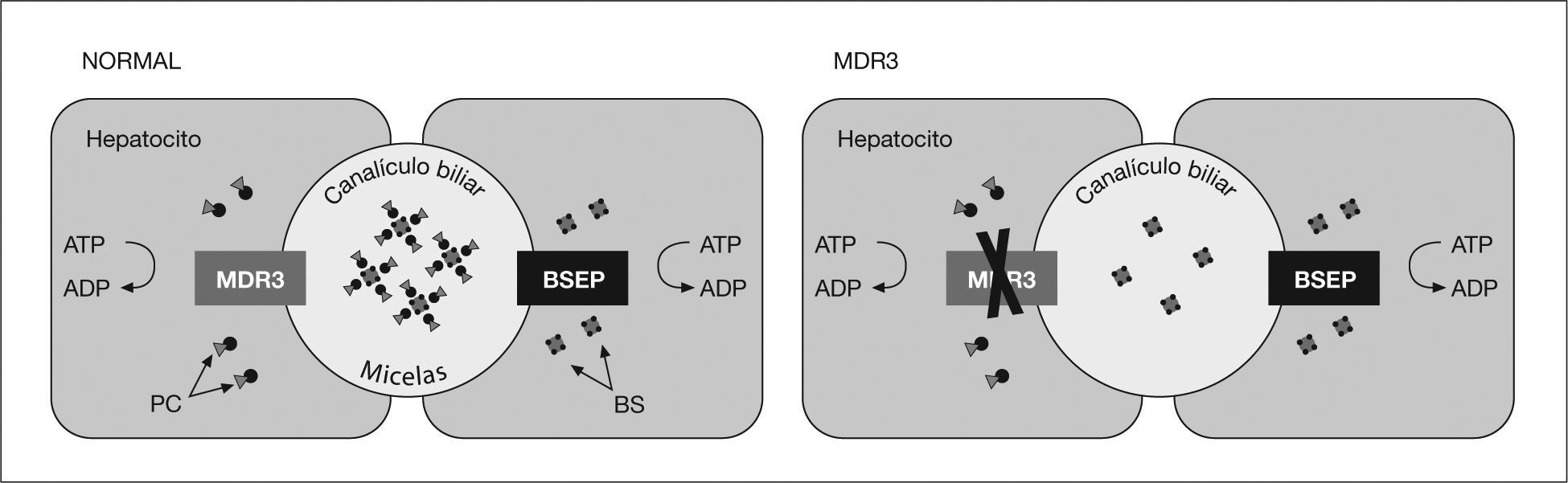
La P-glucoproteína transmembrana (MDR3) interviene en el transporte transmembrana de la fosfatidilcolina al espacio canalicular, que, junto con las sales biliares, forman micelas. En ausencia de fosfatidilcolina, las sales biliares secretadas ejercen su acción detergente en el epitelio canalicular.
La enfermedad es variable según la mutación dé lugar a una proteína truncada o sea una mutación missense con actividad residual2. Los síntomas son ictericia, coluria, acolia, hepatoesplenomegalia y prurito de intensidad variable e inconstante. Menos del 10 % de los casos se inician como colestasis neonatal y la evolución tras el inicio suele ser a colestasis crónica (ictérica o anictérica), hipertensión portal e insuficiencia hepática3,4.
Es frecuente la hipertransaminasemia con cifras muy elevadas de GGT (hasta 25 veces el valor normal), elevación de la fosfatasa alcalina, bilirrubina directa y ácidos biliares, con valores de colesterol en rangos normales. En la biopsia hepática se aprecia proliferación ductal, fibrosis e inflamación portal. El diagnóstico se realiza mediante técnicas de inmunohistoquímica en el tejido hepático. El 66 % de los pacientes no expresan MDR3 en el canalículo biliar, el 22 % muestran una tinción leve y el 12 % expresan MDR3 normalmente. Si la expresión de MDR3 es normal (suele ocurrir en pacientes con mutaciones missense), se recurre al estudio de mutaciones4,5.
El tratamiento es común al de otras colestasis, aporte calórico del 120 % de los requerimientos calóricos, suplementos de vitaminas hidrosolubles y liposolubles (D, E y K) y calcio. Junto a éstos se utilizan fenobarbital, rifampicina, colestiramina y UDCA6. El UDCA mejora los parámetros analíticos, reduce la hepatoesplenomegalia, así como la necesidad de trasplante hepático en algunos pacientes con defecto de MDR37. El trasplante hepático (50 % de los casos) está indicado ante la falta de respuesta clínica al tratamiento. La alteración nutricional grave, el prurito incapacitante y/o el retraso en el crecimiento deben hacer plantear el trasplante antes de que existan signos de insuficiencia hepatocelular. El trasplante permite la normalización de la función sin recidiva de la enfermedad con una supervivencia global del 90 % a los 5 años8.
Las mutaciones en el gen ABCB4 se han relacionado también con colestasis neonatal transitoria, litiasis biliar por cálculos de colesterol, colestasis gravídica, colestasis inducida por anovulatorios y cirrosis idiopática del adulto3,9,10.
En conclusión, el defecto de MDR3 debe sospecharse en trastornos colestásicos y cirrosis hepática idiopática con cifras muy elevadas de GGT. El diagnóstico se establece en el 85 % de los casos mediante técnicas de inmunohistoquímica en la biopsia hepática, aunque en el 15 % habrá que recurrir al estudio de mutaciones. La respuesta al UDCA es variable y la evolución, pese al tratamiento, es, en el 50 % de los casos, a trasplante hepático. Los pacientes afectados de mutaciones tipo missense tienen un inicio más tardío de los síntomas, formas clínicas más leves, menor progresión a cirrosis y mejor respuesta al tratamiento con UDCA.
