







Editado por: Fernando Santos-Simarro. Molecular Diagnostic and Clinical Genetics Unit University Hospital Son Espases. Palma de Mallorca. España
Última actualización: Septiembre 2025
Más datosLos avances en las tecnologías de secuenciación de nueva generación (NGS) han hecho que la detección de las causas moleculares de las enfermedades pediátricas sea cada vez más asequible, accesible y rápida. Si bien hasta hace no mucho la secuenciación del exoma y la secuenciación del genoma solo estaban disponibles para la investigación, ahora se utilizan en la práctica asistencial. La aplicación clínica de la NGS ha planteado muchos retos a la hora de asesorar a las familias sobre la interpretación de los resultados diagnósticos y los hallazgos incidentales, así como las limitaciones técnicas ante resultados genéticos no concluyentes. Dadas las implicaciones de los resultados genéticos en la toma de decisiones médicas, se requiere de un conocimiento especializado de los procedimientos técnicos de los estudios genéticos, sus ventajas y limitaciones, así como del posible impacto (psico)social, legal y ético de los mismos en pacientes, familiares y profesionales sanitarios. Las implicaciones éticas que derivan del hecho de que sean los progenitores quienes otorguen el consentimiento para el estudio de su descendiente y la posible revelación de enfermedades genéticas con opciones terapéuticas limitadas siguen siendo objeto de reflexión. En esta revisión planteamos una pincelada sobre todos estos aspectos, incluyendo las ventajas y las limitaciones de las técnicas actuales de NGS, y mostramos las posibilidades de las soluciones que están por venir.
Advances in next-generation sequencing (NGS) technologies have made the detection of the molecular causes of paediatric diseases increasingly affordable, accessible and rapid. While exome sequencing and genome sequencing were until recently only available for research, they are now used in health care practice. The clinical application of NGS has raised many challenges in genetic counselling for families in terms of the interpretation of test results and incidental findings, as well as technical limitations in the event of inconclusive results. Given the impact of genetic results in clinical decision-making, specialized knowledge is required of the techniques and methods used in genetic studies, their advantages and limitations, and their potential psychosocial, legal and ethical impact on patients, relatives and health care professionals. The ethical implications of parents giving consent to genetic testing in their offspring and the potential disclosure of genetic diseases for which there are limited therapeutic options are still under debate. In this review, we provide an overview of all these aspects, including the advantages and limitations of current NGS techniques, and discuss the possibilities of upcoming solutions.
Aproximadamente 400 millones de personas en todo el mundo están directamente afectadas por una enfermedad rara, siendo el 70% de aparición pediátrica. Actualmente, el tiempo medio desde el inicio de la enfermedad hasta el diagnóstico es de 4,8años1. Reducir este tiempo es importante para mejorar la calidad de vida de estos pacientes y, en algunos casos, puede proporcionar una ventana para la intervención terapéutica.
La mejora de las tecnologías de secuenciación del ADN y la disminución del coste asociado han llevado a la utilización de la secuenciación del exoma completo (WES) y/o del genoma completo (WGS) en el ámbito clínico. Aunque la aplicación de estas tecnologías ha facilitado enormemente la identificación de variantes genéticas asociadas a enfermedades (implicando la creación de consensos para la clasificación de las alteraciones genéticas)2, la tasa de diagnóstico de enfermedades raras sigue siendo inferior al 50%3. ¿Cómo es posible que con toda esta tecnología a nuestro alcance no podamos dar respuesta a tantas familias que acuden a nuestras consultas? En este trabajo pretendemos plasmar el proceso de un estudio genético en pediatría, desde que el paciente entra en la consulta hasta que el estudio termina, con o sin resultado concluyente, revelando las que nos parecen las principales limitaciones y problemáticas que tenemos que abordar y dando una breve pincelada sobre sus posibles soluciones.
Asentando las basesSolemos comparar el genoma humano con una enciclopedia de 23 o 46 volúmenes en la que las letras individuales del texto equivaldrían a los cuatro nucleótidos del ADN, las frases o las palabras, a los genes, y los volúmenes, a los cromosomas.
Así, cuando explicamos las posibles alteraciones que pueden ser responsables de una enfermedad, podemos hablarles a los pacientes de cambios de letras que conllevan alteraciones en una palabra para referirnos a variantes puntuales en la secuencia de ADN y, por tanto, una instrucción incorrecta; o de pérdida de capítulos de libros o de una copia del libro o de una serie de libros (trilogía, pentalogía o «polilogía») cuando queremos hablar de deleciones exónicas, génicas o cromosómicas, respectivamente.
Pero es importante que, como profesionales, tengamos claro que las alteraciones del material genético son un espectro continuo entre cambios a nivel cromosómico a cambios de un único nucleótido. Y que, en función del tipo de alteración esperada (fig. 1), las técnicas más eficientes a emplear pueden ser unas u otras4.

Clasificación de las variantes genéticas. Las variantes genéticas pueden clasificarse en función de la extensión del material genético afectado en alteraciones cromosómicas o alteraciones génicas; o en función del tipo celular implicado en alteraciones somáticas o en línea germinal. También según su efecto, y siguiendo la clasificación del Colegio Americano de Genética Médica (ACMG), en variantes benignas, probablemente benignas, de significado incierto, probablemente patogénicas y patogénicas2.
La Ley de Investigación Biosanitaria 14/2007 especifica que toda persona a la que se le realice un estudio genético ha de recibir siempre, antes y después del mismo, el adecuado asesoramiento genético realizado por personal cualificado.
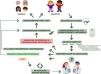
El asesoramiento genético es un proceso comunicativo en el transcurso del cual un profesional especializado proporciona información médica compleja al paciente y/o familiares de forma sencilla respecto a la enfermedad genética, su herencia, el riesgo de recurrencia y las opciones disponibles5 (fig. 2). En este proceso, es imprescindible generar un clima de confianza que permita conocer cuáles son las motivaciones, las expectativas de los pacientes (o progenitores) sobre la consulta, corroborando a la vez qué conocimientos previos poseen. Ese clima brindará la posibilidad de aclarar el propósito, posibles malinterpretaciones y objetivos de una consulta de asesoramiento genético y de un posible estudio genético.

Fases del asesoramiento genético en pediatría. El asesoramiento genético es un proceso centrado en el paciente y su familia. Tras un diagnóstico clínico, el paciente es remitido a la consulta de asesoramiento genético, donde se le exponen las características de su enfermedad, el riesgo, etc.; y tras la firma del consentimiento informado (CI), se realizarán los estudios genéticos pertinentes. Los resultados deben ser interpretados por personal especializado, pudiendo encontrarnos ante estas posibilidades: variantes (probablemente) patogénicas, variantes (probablemente) benignas y variantes de significado incierto (VUS). Asimismo, dependiendo de lo que se eligiera durante la firma del CI, se deberá informar sobre los hallazgos incidentales.
Los profesionales implicados en el proceso de asesoramiento genético pretenden ayudar al paciente y/o familiares a6:
- •
Entender los conceptos médicos de la enfermedad, incluyendo el diagnóstico, el curso, las causas y el manejo clínico.
- •
Saber cómo la herencia contribuye a la patología y el riesgo de recurrencia u ocurrencia en la familia.
- •
Entender las alternativas para reducir el riesgo de ocurrencia, recurrencia o transmisión.
- •
Elegir la actuación más apropiada en función del riesgo, las opciones disponibles y los principios éticos y/o religiosos.
- •
En caso de disponer de estudio genético, entender las implicaciones, las ventajas y las limitaciones del estudio y los resultados que se pueden derivar, así como sus consecuencias.
- •
Asesorar y apoyar psicoemocionalmente para promover decisiones informadas adaptándose de la mejor manera al riesgo o condición genética.
- •
El objetivo final del proceso de asesoramiento genético es facilitar la toma de decisiones de acuerdo con los valores y creencias de la familia y actuar en concordancia con la decisión tomada.
- •
La consulta de asesoramiento genético se diferencia de otras consultas clínicas en que no solo se enfoca en quien acude a la consulta, sino que también tiene implicaciones para su familia.
Durante la primera consulta se exploran los antecedentes personales y familiares con la información que el paciente o la familia proporcione, integrándolos en un árbol genealógico (pedigrí) siguiendo la nomenclatura internacional7,8 (fig. 3), con el objetivo de identificar la probabilidad de presentar una predisposición heredable a alguna enfermedad. Se construye una genealogía de al menos tres generaciones, lo que nos permite visualizar a los integrantes de la familia, señalando a enfermos y sanos. La información incluye la edad actual, el estado de salud, la edad en el momento de la muerte y la causa de la muerte, y los diagnósticos médicos con exposiciones medioambientales relacionadas. Es probable que durante su elaboración obtengamos indirectamente información de las relaciones personales entre los distintos miembros de la familia9.

Ejemplos de árboles genealógicos (pedigrís). Los símbolos del pedigrí se refieren al género: los cuadrados representan varones; los círculos, mujeres; los triángulos, abortos espontáneos; los triángulos cruzados, interrupción legal/voluntaria del embarazo; los rombos, personas con género desconocido o no binario; los símbolos cruzados indican que la persona ha fallecido, y la letra «P» indica gestación en curso. La flecha señala la persona consultante o la persona por la que se consulta, y la flecha junto a una letra «P» indica al probando. Los números romanos hacen referencia a la generación, y los números arábigos, al individuo. AMAB: asignado hombre al nacer (assigned male at birth).
A) Familia con antecedentes de abortos en varios de los miembros. La consultante desea conocer su riesgo de tener un nuevo descendiente enfermo. A la vista del pedigrí, se plantea que lo más probable es que sea portadora de una translocación equilibrada, pudiendo una nueva gestación conllevar a un nuevo aborto, a un descendiente con enfermedades congénitas debido a heredar la translocación desequilibrada, a un descendiente sano portador de la misma translación equilibrada o a un descendiente con una dotación cromosómica sin alteración. Se le ofrecerá la posibilidad de realizar un cariotipo.
B) Ante un antecedente familiar de acondroplasia, se consulta sobre el riesgo de recurrencia en un embarazo en curso. Dada la herencia autosómica dominante de dicha enfermedad, y que ninguno de los progenitores la manifiesta, se exponen a la pareja la posibilidad de mosaicismo gonadal, y los riesgos y beneficios de una amniocentesis para la detección de la variante familiar.
C) A la vista del árbol familiar, se infiere que los más probable es que ambos progenitores sean portadores de la variante causal de la hipoacusia. Por tanto, el riesgo a priori del nuevo descendiente de presentar la enfermedad es del 25%. Se ofrece la posibilidad de estudio genético dirigido a ambos progenitores.
La evaluación de la genealogía permite discriminar los factores de riesgo genéticos (hereditarios) de los factores ambientales (estilo de vida, exposición a tóxicos, etc.), y además es de gran utilidad como herramienta de educación en la promoción en salud y la prevención de enfermedades. Tenemos que ser conscientes de que, en muchas ocasiones, esta recolección de información tiene el sesgo del recuerdo de las personas que acuden a la consulta y que pueden confundir edades de diagnóstico o fallecimiento e, incluso, diagnósticos clínicos. En caso de ser relevantes para la orientación del asesoramiento, es recomendable indicarles que busquen esa información (o autorización por parte de familiares implicados para que nosotros podamos consultar historiales clínicos) y acudir a una nueva consulta.
Con esta información, se establece el posible modo de herencia y se informan de los riesgos de recurrencia (probabilidad de que los progenitores tengan otro descendiente con la misma enfermedad que el consultante) y de transmisión (probabilidad de que, en un futuro, la persona que consulta transmita la enfermedad a su descendencia)9.
Asimismo, se informa sobre en qué consisten los estudios genéticos que pudieran estar disponibles para la enfermedad, sobre la probabilidad de presentar una predisposición heredable, sobre los posibles resultados (tabla 1), sobre la probabilidad de transmisión, de recurrencia, las implicaciones médicas, las medidas de prevención y de diagnóstico precoz, las opciones reproductivas, etc. Se describe el procedimiento, potenciales beneficios y limitaciones.
Posibles resultados a obtener tras un estudio genético orientado a la patología de consulta, con algunos de sus beneficios y riesgos. Se considera un resultado positivo cuando el estudio genético identifica una variante genética que probablemente sea la causa de la enfermedad (patogénica). Un estudio genético es no informativo o no concluyente cuando no revela variante(s) patogénica(s) o probablemente patogénica(s) que explique(n) la causa de la enfermedad. Se considera que el estudio genético es (verdadero) negativo cuando la persona estudiada no presenta la variante familiar
| Resultados | Beneficios | Riesgos |
|---|---|---|
| Positivos (variante patogénica o probablemente patogénica) | • Confirmación del diagnóstico• Intervención precoz• Identificación de familiares a riesgo• Reduce la incertidumbre• Comportamiento saludable• Orientación reproductiva | • Impacto psicosocial• Dinámica familiar• Incertidumbre - Penetrancia• Discriminación• Confidencialidad• Etiquetador y justificación de comportamientos desde la enfermedad• Sensación de culpabilidad |
| No informativo o no concluyenteVariante de significado incierto | • Clarificación por medio de investigaciones en el futuro• Reforzar el comportamiento saludable y el seguimiento de las recomendaciones de los clínicos de referencia | • Incertidumbre• Falsa seguridad• Mala adherencia a las medidas preventivas• Ansiedad y frustración• Enfado• Necesidad de nuevos estudios genéticos |
| «Verdadero» negativo(variante familiar conocida) | • Alivio emocional• Evita intervenciones o pruebas innecesarias• Implicaciones en descendencia | • Reacciones psicológicas «survivors guilt»• Mala adherencia a las medidas preventivas de la población en general (casos oncológicos) |
Existe la creencia de que acudir a una consulta de asesoramiento implica la toma de muestra para el estudio genético, cuando en realidad el objetivo es obtener información y educar sobre los riesgos, las implicaciones de los resultados y las medidas de prevención y manejo, de manera no directiva. Dado que el asesoramiento genético no siempre desemboca en la recomendación de pruebas genéticas, es importante que el clínico remitente informe a los pacientes de que se les deriva para determinar si las pruebas genéticas serían apropiadas, en lugar de para someterse a ellas. Si la única expectativa de la visita es que se realice una prueba genética y esto no ocurre, los pacientes pueden sentirse frustrados.
Por otra parte, en caso de estar disponible, el estudio genético es optativo y voluntario; respetando su autonomía, los pacientes o sus responsables legales pueden decidir realizar el estudio genético o declinarlo una vez obtenida toda la información. Es frecuente que familias que estaban decididas a que se realizara el estudio genético prefieran postergarlo tras la consulta de asesoramiento genético, ya sea para compartir la información con otros familiares (muy frecuente cuando a consulta acude uno solo de los progenitores) o con el clínico de referencia, o para reflexionar sobre las implicaciones y las posibilidades que estos estudios ofrecen. En el caso de estudios de secuenciación masiva es especialmente compleja la toma de decisión por parte de los progenitores, por la comprensión de las implicaciones tanto a nivel de salud10 como éticas11.
En caso de que el paciente o la familia muestren interés en su realización, se explica quién es la persona candidata al estudio, la persona más «informativa» de la familia, que no siempre tiene que ser quien consulta. Es importante aclarar por qué se estudia primero a una u otra persona, su implicación en la relevancia de los resultados. Una vez ante la persona candidata y que ha aceptado realizarse el estudio, se explica y entrega el consentimiento informado, que debe firmarse por ambos progenitores o tutores legales, en caso de menores de 12años; o por el menor capaz y un progenitor o tutor legal, en caso de personas con más de 12años; o por el consultante si es mayor de edad (art.5.3 del Real Decreto 1090/2015; art.20.1 de la Ley 14/2007). Existen unas recomendaciones sobre los elementos imprescindibles que debiera contener un consentimiento informado para los estudios basados en secuenciación masiva12.
Los estudios genéticos se realizan, generalmente, sobre una muestra de sangre extraída a tal efecto una vez otorgado el consentimiento. Insistimos, la decisión sobre hacer o no el estudio genético es personal y siempre ha de tomarla el paciente o los responsables legales adecuadamente informados.
Estudio o test genéticoDebemos tener presente que existen enfoques diferentes para las pruebas genéticas, desde la secuenciación de un solo gen hasta de todo el genoma, gracias a la tecnología de secuenciación de nueva generación (next-generation sequencing [NGS]). De hecho, la NGS puede utilizarse para secuenciar regiones concretas del genoma o grupos de genes (lo que se conoce comúnmente como paneles de genes), secuenciar todas las regiones codificantes o exones (WES) o secuenciar todas las regiones codificantes, no codificantes e intergénicas (WGS). Cada una de estas estrategias presenta ventajas y limitaciones que deben tenerse en cuenta a la hora de seleccionar la prueba más adecuada para el paciente (tabla 2).
Ventajas y limitaciones de cada una de las técnicas de NGS, así como las indicaciones en las que es óptimo usar cada una de ellas
| Técnica | Indicaciones | Ventajas | Limitaciones |
|---|---|---|---|
| Gen único | Sospecha de enfermedad monogénica con características bien definidasPruebas de portadores y cosegregación en las familias | Precio asequibleNinguno o pocos hallazgos incidentales | Puede que no incluya genes asociados recientemente al fenotipoNo permite identificar CNV ni SV |
| Panel de genes | Sospecha de una enfermedad con muchos genes asociadosPatologías con fenotipos similaresEnfermedades monogénicas con sospecha de alteración en mosaico | Menos hallazgos incidentales en comparación con la WES y WGSPueden capturar regiones específicas no codificantes (reguladoras o intrónicas) y regiones complejasPermite identificar variantes en mosaicoAnálisis de datos sencillo y poca cantidad de datos para almacenar | Normalmente se estudia solo el índice o probando, lo que requiere pasos adicionales para confirmar la herenciaPuede que no incluya genes asociados recientemente al fenotipo y no detecte variantes en genes novelesNo permite identificar CNV ni SVNo captura regiones repetitivas o pseudogenes |
| Exoma (WES) | Pruebas previas no identifican la variante causalFenotipo que no se ajusta a un síndrome bien descritoNecesidad de una prueba amplia y rápida (p.ej., prenatales, unidad de cuidados intensivos…)Panel de genes no disponible para fenotipo | Permite realizar una amplia evaluación de la mayoría de los genes codificantes de proteínasPermite identificar variantes en nuevos genes no asociados previamente a la enfermedadLos datos pueden ser reanalizadosPermite identificar CNV, aunque de forma limitada | Puede detectar hallazgos incidentalesNo captura regiones no codificantes (reguladoras o intrónicas profundas)No captura regiones repetitivas o pseudogenesNormalmente la profundidad media no es suficiente para detectar variantes en mosaico (×100)Múltiples tipos de librerías para capturar el exoma, lo que significa que no todos los estudios son equivalentes |
| Genoma (WGS) | Pruebas previas no identifican la variante causalFenotipo que no se ajusta a un síndrome bien descritoEl fenotipo sugiere un mecanismo de expansión | Análisis no sesgado de todo el genomaPermite identificar variantes tanto en regiones codificantes como no codificantesPermite identificar variantes en nuevos genes no asociados previamente a la enfermedadLos datos pueden ser reanalizadosPermite identificar CNV y SVPermite capturar regiones repetitivas o pseudogenes | Coste elevadoPuede detectar hallazgos incidentalesLa profundidad media no es suficiente para detectar variantes en mosaico (×30-50)Análisis de datos complejo y mucha cantidad de datos para almacenarRequiere considerables recursos informáticos y de redFalta de bases de datos públicas a nivel de población para su filtrado e interpretación |
CNV: variantes de número de copia; SV: variantes estructurales; WES: secuenciación exoma completo; WGS: secuenciación genoma completo.
La secuenciación de un gen seleccionado puede ser una prueba genética adecuada para una patología en la que las mutaciones en un gen concreto son la principal causa de la enfermedad. Sería, por ejemplo, el caso de los pacientes con acondroplasia, donde el 99% presentan la variante patogénica c.1138G>A en heterocigosis en el gen FGFR3 y un 1% la variante c.1138G>C13; o el de los pacientes con fibrosis quística, que presentan alteraciones bialélicas en el gen CFTR14.
Sin embargo, debido a la heterogeneidad genética y fenotípica de la mayoría de las enfermedades pediátricas hereditarias, la secuenciación de un único gen no suele ser el enfoque más eficiente o eficaz.
Otro uso de la secuenciación de un solo gen o de la secuenciación dirigida es para analizar a los miembros de la familia en busca de la variante específica identificada previamente como causante de la enfermedad en esa familia.
Las pruebas de gen único también pueden realizarse para la detección de portadores en la pareja reproductiva de un paciente con una enfermedad autosómica recesiva conocida o portador para ella. La determinación del estado de portador de ambos miembros de la pareja proporciona una evaluación más precisa del riesgo para su descendencia15.
Estos estudios pueden realizarse bien por secuenciación tradicional (secuenciación Sanger) o por NGS.
Paneles de genesMuchas de las enfermedades pediátricas presentan heterogeneidad genética y fenotípica, con más de un gen causante responsable de una enfermedad concreta, o una serie de fenotipos clínicos derivados de alteraciones en un único gen. Por ejemplo, en el caso de las hipoacusias16, la epilepsia17 o las displasias esqueléticas18 se han descrito cientos de genes implicados como causantes de la enfermedad. En estos casos es beneficioso secuenciarlos utilizando un panel de genes, que se basa en el uso de la tecnología NGS para seleccionar o capturar exones y secuenciar las regiones o genes seleccionados. Tras la secuenciación, se requieren análisis bioinformáticos para identificar las posibles variantes causales.
Una ventaja importante de este enfoque es la capacidad de secuenciar simultáneamente muchos genes sin tener que realizar el estudio de un gen tras acabar el del anterior. Además, por lo general tienen una alta profundidad de lectura (la misma posición es secuenciada múltiples veces), lo que permite identificar alteraciones en mosaico.
Las desventajas incluyen un mayor coste en comparación con la secuenciación dirigida o de un solo gen, la identificación de variantes de significado incierto y la posibilidad de descubrir hallazgos incidentales (véase más adelante).
Secuenciación de exoma (WES) y secuenciación de genoma (WGS)Dado que el coste de la secuenciación disminuye continuamente, tanto la WES como la WGS se utilizan cada vez más como pruebas diagnósticas19,20. Ambos métodos pueden identificar de forma exhaustiva todas las variantes genéticas en regiones codificantes. En el caso de la WGS, también se pueden identificar las variantes genéticas en secuencias no codificantes (variantes en el promotor, intergénicas, en zonas reguladoras…). Estos métodos tienen la ventaja de descubrir nuevos genes causantes de la enfermedad y de ampliar el espectro fenotípico de los genes ya conocidos. Por último, es importante tener presente que, en función del pipeline bioinformático, además de las alteraciones en la secuencia, también podremos analizar alteraciones cromosómicas a gran escala, como deleciones o duplicaciones21.
El WES presenta ciertas limitaciones. Por lo general, no detecta variantes intrónicas (a menos que flanqueen inmediatamente un exón diana), tampoco detecta expansiones de repeticiones de trinucleótidos ni anomalías de metilación (estas últimas tampoco son detectables por WGS), y es posible que solo detecte de forma limitada las variantes de número de copia19.
Al igual que las pruebas basadas en paneles, las pruebas WES y WGS requieren sofisticados análisis bioinformáticos (más en este caso dado que el número de variantes que se identifican es aún mayor) para detectar variantes potencialmente patogénicas de entre las variantes de ADN identificadas mediante este proceso.
Para el descubrimiento de nuevos genes es útil secuenciar a los miembros de la familia (exoma/genoma trío)19, y en la práctica pueden ser necesarios resultados de diferentes familias para proporcionar pruebas sólidas de un nuevo gen patogénico. Las desventajas de WES y WGS incluyen costes más elevados, mayores demandas de almacenamiento de datos, plazos de entrega más largos, necesidad de personal experto en análisis bioinformáticos y, por último, la posibilidad de descubrir hallazgos genéticos incidentales (véase más adelante).
Pruebas de detección de alteraciones estructuralesLa variación genómica estructural se define como cambios genómicos >1kb (kilobase) que pueden ser desequilibrados (deleciones, inserciones y duplicaciones, también llamados CNV) o equilibrados (translocaciones e inversiones)22.
Si bien, como hemos mencionado, algunas aproximaciones bioinformáticas permiten la detección de algunas de estas alteraciones en los estudios de NGS, en ocasiones se requieren métodos o tecnologías específicas que incluyen la hibridación fluorescente in situ (FISH) y la amplificación de sonda dependiente de ligación múltiple (MLPA), cuando se conoce la región que puede estar afectada, o la hibridación genómica comparativa (CGH), los chips de polimorfismo de nucleótido único (SNP), cuando son estudios a genoma completo.
Las variantes estructurales representan una parte significativa de la «causalidad ausente» en las enfermedades pediátricas, cuando los estudios de secuenciación no permitían identificar (o no buscaban) este tipo de alteraciones23. Es importante tener presente que el análisis genético completo de los pacientes con enfermedades hereditarias debería incluir tanto la secuenciación como el análisis de las CNV.
Interpretando los resultados de un estudio genéticoLa NGS genera miles de variantes de secuencia potencialmente asociadas a enfermedades. A medida que aumenta el número de genes secuenciados, es necesario evaluar un mayor número de variantes. Los análisis bioinformáticos son un paso fundamental para identificar las realmente causantes de enfermedad. Además, proporcionar una descripción precisa de los fenotipos clínicos, según la codificación HPO (https://hpo.jax.org/app/), es útil cuando se envían muestras de pacientes para estudios genéticos. Estas descripciones pueden ayudar a priorizar las variantes identificadas24.
El Colegio Americano de Genética Médica (American College of Medical Genetics [ACMG]) ha proporcionado una serie de directrices para evaluar la patogenicidad de las variantes utilizando la frecuencia poblacional, los análisis computacionales (insilico), los datos funcionales, así como los datos clínicos y la segregación familiar, para asignarles una clasificación de patogénica, probablemente patogénica, de significado incierto, probablemente benigna o benigna2. ClinGen (Clinical Genome Resourcehttps://clinicalgenome.org/), GenCC (Gene Curator Coalition, https://search.thegencc.org/), Franklin (https://franklin.genoox.com/clinical-db/home), ClinVar (www.ncbi.nlm.nih.gov/clinvar) y LOVD (www.lovd.nl) proporcionan recursos adicionales para la interpretación de variantes.
Variantes de significado inciertoLas variantes de significado incierto (VUS) pueden tener algunas características de las variantes causantes de enfermedades, pero no hay pruebas suficientes para apoyar una clasificación patogénica ni una clasificación benigna. Un ejemplo de VUS es una variante en un gen conocido como causante de enfermedad que se encuentra raramente en la población, pero sin datos funcionales que apoyen su patogenicidad. Otras pruebas que apoyarían la patogenicidad podrían incluir pruebas funcionales o pruebas de segregación con enfermedad en miembros de la familia, o en otra familia con la misma variante y la misma afección.
Las VUS pueden reclasificarse con el tiempo25, por lo que es importante que la persona que se somete a las pruebas se mantenga en contacto con el clínico que solicitó o realizó las pruebas genéticas para asegurarse de recibir actualizaciones si se conoce nueva información sobre la variante.
Hallazgos inesperadosLos estudios de secuenciación masiva pueden identificar variantes responsables de patologías diferentes a la enfermedad por la que se consulta. Estos resultados no requeridos se han dividido en hallazgos secundarios y hallazgos incidentales. Si bien ambos son variantes (probablemente) patogénicas no relacionadas con la cuestión clínica inicial, los hallazgos secundarios se refieren a aquellas variantes localizadas en genes que son buscados activamente por el laboratorio clínico y cuya búsqueda se basa en la lista propuesta por el ACMG26, e incluye genes seleccionados por estar asociados con enfermedades que disponen de una prueba genética clínica fiable y una intervención o tratamiento eficaces. Por otro lado, los hallazgos incidentales incluyen variantes (probablemente) patogénicas no relacionadas con la indicación clínica primaria que se identifican por casualidad durante el análisis genético27.
No hay un claro consenso sobre si los hallazgos genéticos incidentales deben o no revelarse automáticamente a los pacientes28,29. Este aspecto es aún más difícil de abordar cuando el estudio genético se va a realizar en niños11. Además, no podemos perder de vista que, si se realiza la secuenciación del exoma trío, estas variantes no solicitadas, cuando se encuentran en el paciente, podrían incluirse en el informe y añadir información relativa a la herencia para las variantes, y por lo tanto, tiene el potencial de diagnosticar a un progenitor al mismo tiempo que a su descendiente. Incluso si los miembros de la familia no se someten a la secuenciación, la identificación de un hallazgo incidental en un niño puede tener implicaciones para toda la familia, ya que se pueden recomendar pruebas en cascada para los miembros no afectados. Esta implicación de los resultados de los estudios genéticos de los niños en sus progenitores podría tener un efecto en la decisión de estos sobre la realización o no del estudio genético a su descendiente11.
Consulta post-testCon todas estas opciones de resultados (desde las variantes patogénicas hasta las VUS o las inesperadas), lo óptimo sería poder realizar esta consulta junto con el clínico de referencia del paciente, que será quien se hará cargo de su seguimiento.
En esta sesión se expone al paciente y/o a los progenitores que los resultados ya están disponibles y se les consulta de nuevo por su interés en conocerlos. Se confirma si desean ser informados de los hallazgos incidentales en caso de que así lo indicaran en la consulta pre-test. Se aclara cualquier duda que hayan tenido durante el tiempo de espera hasta la llegada de los resultados.
Se les informa de los resultados, se explican en su contexto, sus implicaciones médicas, cómo se hereda la enfermedad, el posible riesgo de otros familiares, y opciones reproductivas. Se aclaran las posibles dudas y preocupaciones que surjan en la consulta. Se indican las recomendaciones de derivación a los especialistas según la alteración genética para poder hacer un seguimiento adecuado al riesgo y se remite al paciente (y a los familiares) a los especialistas correspondientes.
Además, podremos tener que exponer y aclarar los hallazgos incidentales, y sus implicaciones, tanto en el consultante como en el resto de miembros de la familia.
Habitualmente esta sesión implica algo más que el suministro de información médica y suele centrarse en ayudar a las familias a afrontar las consecuencias emocionales, psicológicas, médicas, sociales y económicas de los resultados de la prueba. En particular, se abordan cuestiones psicológicas como la negación, la ansiedad, la ira, el dolor o la culpabilidad, y cuando es necesario se ofrecen derivaciones para un asesoramiento psicosocial en profundidad.
Se entrega un informe donde se resumen los motivos de consulta, los estudios genéticos realizados y los hallazgos30, así como recomendaciones y material para compartir con los familiares o sobre recursos comunitarios y grupos de apoyo o asociaciones de pacientes (si hubiera). Si es posible, se entregan datos de contacto para establecer una relación abierta en caso de más preguntas o consultas.
Si la prueba genética es positiva, se puede considerar la realización de pruebas a otros familiares del individuo, por lo que se ofrece la posibilidad de derivar a otros miembros de la familia para la evaluación del riesgo.
Limitaciones y próximos pasosEntre el 50% y el 60% de los individuos con una presunta enfermedad mendeliana siguen sin ser diagnosticados tras la realización de pruebas genéticas exhaustivas (aunque la tasa de diagnóstico varía en función de la patología).
Son varios los factores que contribuyen a no alcanzar este diagnóstico genético confirmatorio:
- •
La base genética de muchas enfermedades mendelianas sigue siendo desconocida.
- •
En el caso de enfermedad para la que se conoce el gen o los genes subyacentes, es posible que la prueba solicitada no interrogue al gen o genes apropiados (por ejemplo, paneles de un solo gen o de varios genes), al tipo o tipos de variantes (por ejemplo, expansión de tripletes) o a alteraciones epigenéticas (por ejemplo, estado de metilación).
- •
Las limitaciones técnicas pueden dificultar la identificación de una variante patogénica (por ejemplo, la detección de una CNV a partir de un WES).
- •
Puede que no haya suficiente información para interpretar la patogenicidad de una variante. Este problema se ve agravado por el hecho de que la interpretación de las variantes por parte de los laboratorios de diagnóstico puede variar sustancialmente debido a las diferencias en la ponderación de las pruebas de patogenicidad de una variante31, aunque la estandarización de la clasificación y los esfuerzos por compartir datos deberían mitigar este efecto32,33.
- •
La penetrancia incompleta y los retos asociados a distinguir si un fenotipo se debe a alelos de gran efecto o es el resultado de patrones de herencia complejos (por ejemplo, digénicos u oligogénicos) dificultan la identificación de la etiología molecular de un fenotipo.
- •
La tasa de diagnóstico ha dependido históricamente de la profundidad de la información fenotípica disponible en la adjudicación de variantes34. Aunque no existen directrices generales para el fenotipado sistemático, las propuestas de utilizar términos de ontología de fenotipos humanos y paquetes de fenotipos proporcionarían un estándar para el intercambio de fenotipos entre laboratorios, médicos e investigadores24.
- •
También debe reconocerse que las explicaciones no genéticas, como las infecciones o la exposición a los tóxicos, pueden ser responsables de algunas de estas enfermedades.
Para solventar algunas de estas limitaciones, distintos laboratorios se plantean realizar reanálisis de los estudios NGS y/o reinterpretaciones de las variantes identificadas, bien a solicitud del clínico remitente o incluso del paciente, por aparecer nuevos familiares enfermos o nuevas manifestaciones clínicas35. Incluso hay quienes abogan por realizar estas revisiones de manera sistemática con periodicidades que oscilan entre uno36 y tres años37. En el caso de los estudios sistemáticos, sería importante valorar las implicaciones éticas, económicas, jurídicas y (psico)sociales que estas pudieran tener, tanto para pacientes y familias, como para los profesionales sanitarios involucrados38.
Por último, no podemos perder de vista las prometedoras nuevas tecnologías, como la secuenciación de lecturas largas (long-read sequencing)39 o el mapeo óptico del genoma (optical genome mapping)40, que ayudarán a dar respuesta a algunos de los pacientes que aún permanecen sin diagnóstico.
FinanciaciónNinguna.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
