






Las anomalías vasculares (AV) son alteraciones de la vascularización que suelen aparecer en el feto, al nacimiento o en la primera infancia. Pueden causar dolor crónico, disfunción motora, trastornos cosméticos, coagulopatía y son ocasionalmente mortales, pero en todos los casos empeoran la calidad de vida del niño y su familia. Existen hasta 150 subtipos diferentes descritos. Pueden afectar arterias, capilares, venas, linfáticos o a una combinación de ellos. Pueden estar asociadas a malformaciones adicionales y causan con frecuencia hipertrofia o hipotrofia musculoesquelética y de partes blandas. Pueden localizarse en cualquier parte del cuerpo, invadir cualquier tejido y afectar a la función de diversos órganos. Las prevalencias de los diferentes subtipos varían enormemente, de 1/20 a 1/1.000.000. Los subtipos considerados enfermedades raras (incidencia inferior a 1/2.000) siguen afectando a más de 500.000 personas en la Unión Europea (UE).
La diferenciación entre tumores vasculares y malformaciones vasculares es crítica, especialmente cuando se trata de pacientes pediátricos. Las dos entidades representan patologías completamente diferentes, aunque agrupadas bajo el paraguas común del término AV.
El algoritmo diagnóstico utilizado en la evaluación clínica de las AV debe basarse en una historia clínica completa y un examen físico detallado.
El futuro del conocimiento en este campo se basará sobre todo en los hallazgos genéticos y en las innovaciones terapéuticas. Nuevas moléculas y sus indicaciones están siendo exploradas, con destino a reducir la agresividad de tratamientos previos y de dar esperanza y calidad de vida a los pacientes sin respuesta a los tratamientos convencionales.
Vascular anomalies are changes in vascularization that usually appear in the foetal stage, at birth or in early childhood. They can cause chronic pain, motor impairment, cosmetic changes or coagulopathy and may be fatal in some cases, but in every case they have a negative impact on the quality of life of the child and the family. Up to 150 different subtypes have been described. They can involve arteries, capillaries, veins, lymphatic vessels or a combination thereof. They may be associated with additional malformations and frequently cause musculoskeletal and soft tissue hypertrophy or hypotrophy. They can develop anywhere in the body, invade any tissue and affect the function of various organs. The prevalence of the different subtypes varies greatly, from 1/20 to 1/1 000 000. Subtypes considered rare diseases (incidence <1/2000) continue to affect more than 500 000 people in the European Union.
Differentiating between vascular tumours and vascular malformations is critical, especially in paediatric patients. They are completely different diseases, although they are often grouped under the umbrella term of vascular anomalies.
The diagnostic algorithm used in the clinical evaluation of vascular anomalies should be based on a thorough history-taking and detailed physical examination.
Future knowledge in this field will be based above all on genetic findings and therapeutic innovations. New molecules and their indications are being explored with the aim of reducing the aggressiveness of previous treatments and increasing the life expectancy and quality of life of patients who do not respond to conventional treatments.
Las anomalías vasculares (AV) son alteraciones de la vascularización que suelen aparecer en el feto, al nacimiento o en la primera infancia. Pueden causar dolor crónico, disfunción motora, trastornos cosméticos, coagulopatía y son ocasionalmente mortales, pero en todos los casos empeoran la calidad de vida del niño y su familia. Existen hasta 150 subtipos diferentes descritos. Pueden afectar arterias, capilares, venas, linfáticos o a una combinación de ellos. Pueden estar asociadas a malformaciones adicionales y causan con frecuencia hipertrofia o hipotrofia musculoesquelética y de partes blandas. Pueden localizarse en cualquier parte del cuerpo, invadir cualquier tejido y afectar a la función de diversos órganos. Las prevalencias de los diferentes subtipos varían enormemente, de 1/20 a 1/1.000.000. Los subtipos considerados enfermedades raras (incidencia inferior a 1/2.000) siguen afectando a más de 500.000 personas en la Unión Europea (UE) (figs. 1 y 2).


Los intentos de diagnóstico y clasificación de las AV tienen una larga historia que comienza con el uso indiscriminado de epónimos y términos descriptivos como «mancha de vino de Oporto», «angioma en fresa», «angioma cavernoso», entre otros.
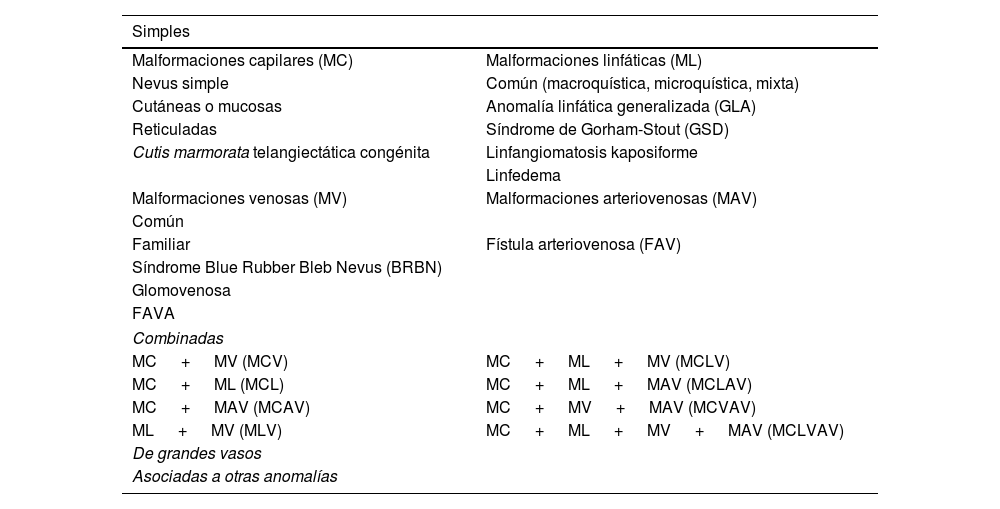
Con la rápida proliferación de esta nomenclatura poco científica, se hizo evidente la necesidad de desarrollar una clasificación biológica de las AV. Esta fue introducida por John Mulliken en 19821 y constituyó la base de la posterior clasificación de la Sociedad Internacional para el Estudio de las Anomalías Vasculares (ISSVA)2,3tablas 1 y 2.
Tumores vasculares
| Tumores benignos | Localmente agresivos | Malignos |
|---|---|---|
| Hemangioma infantil | Hemangioendotelioma kaposiforme | Angiosarcoma |
| Hemangioma congénito | Hemangioendotelioma retiforme | Hemangioendotelioma epitelioide |
| Rápidamente involutivo (RICH) | Angioendotelioma papilar intralinfático | |
| No involutivo (NICH) | Tumor de Dabska | |
| Parcialmente involutivo (PICH) | Hemangioendotelioma compuesto | |
| Tufted angioma | Hemangioendotelioma miogénico | |
| Hemangioma de células fusiformes | Hemangioendotelioma polimorfo | |
| Hemangioma epiteliode | Hemangioendotelioma no específico | |
| Granuloma piogénico | Sarcoma de Kaposi | |
| Otros |
Malformaciones vasculares
| Simples | |
|---|---|
| Malformaciones capilares (MC) | Malformaciones linfáticas (ML) |
| Nevus simple | Común (macroquística, microquística, mixta) |
| Cutáneas o mucosas | Anomalía linfática generalizada (GLA) |
| Reticuladas | Síndrome de Gorham-Stout (GSD) |
| Cutis marmorata telangiectática congénita | Linfangiomatosis kaposiforme |
| Linfedema | |
| Malformaciones venosas (MV) | Malformaciones arteriovenosas (MAV) |
| Común | |
| Familiar | Fístula arteriovenosa (FAV) |
| Síndrome Blue Rubber Bleb Nevus (BRBN) | |
| Glomovenosa | |
| FAVA | |
| Combinadas | |
| MC + MV (MCV) | MC + ML + MV (MCLV) |
| MC + ML (MCL) | MC + ML + MAV (MCLAV) |
| MC + MAV (MCAV) | MC + MV + MAV (MCVAV) |
| ML + MV (MLV) | MC + ML + MV + MAV (MCLVAV) |
| De grandes vasos | |
| Asociadas a otras anomalías | |
M: malformación; C: capilar; V: venosa; L: linfática; AV: arteriovenosa; MCLVAV: malformación capilar linfática venosa y arteriovenosa.
El futuro del conocimiento en este campo se basará sobre todo en los hallazgos genéticos y en las innovaciones terapéuticas. Se han identificado ya cerca de 50 mutaciones en distintas AV y la aplicación del Whole Exome Secuence (WES) a los tejidos afectos ampliará enormemente este catálogo genético (figs. 3, 4 y 5).



La edad se considera un factor determinante en la evaluación clínica de las AV ya que el feto, recién nacido, bebé, niño y adulto pueden presentarse con diferentes signos y síntomas estrechamente relacionados con su periodo de vida.
El algoritmo diagnóstico utilizado en la evaluación clínica de las AV debe basarse en una historia clínica completa y un examen físico detallado. Los aspectos más importantes de la historia son si la lesión estaba presente al nacer (malformaciones vasculares) y la tasa de crecimiento (proporcional o desproporcionada al crecimiento del niño). La excepción viene representada por los hemangiomas congénitos: hemangioma congénito rápidamente involutivo (RICH) y hemangioma congénito no involutivo (NICH). Ambos proliferan exclusivamente en el periodo fetal.
La diferenciación entre hemangiomas y malformaciones vasculares congénitas es crítica, especialmente cuando se trata de pacientes pediátricos. Las dos entidades representan patologías completamente diferentes, aunque agrupadas bajo el paraguas común del término AV. Las malformaciones son errores de morfogénesis vascular en el contexto de mosaicismos somáticos y más raramente, mutaciones en línea germinal. Los tumores vasculares pueden ser benignos (la gran mayoría), localmente agresivos o malignos (angiosarcoma y hemangioendotelioma epitelioide).
Tumores vascularesHemangioma de la infanciaLos hemangiomas de la infancia (HI) son las formas más comunes de tumores vasculares pediátricos que tienen una etiología, presentación, pronóstico y tratamiento específicos. Estas lesiones son proliferativas y, normalmente, no están presentes al nacer, apareciendo en las primeras semanas de vida, con un crecimiento autolimitado, caracterizado por un proceso de proliferación rápida y regresión lenta. La mayoría han sufrido atrofia e involución completa, con leve residuo fibroadiposo y telangiectasia residual, a la edad de 10 años. Histológicamente, presentan tinción positiva para el transportador de glucosa-1 (GLUT-1) durante todas las etapas de crecimiento e involución, lo que facilita su diagnóstico, sobre todo en tumores extracutáneos profundos.
Hemangiomas congénitosLos HI deben diferenciarse de los hemangiomas congénitos, ya que su tratamiento es diferente. Los hemangiomas congénitos se desarrollan completamente en el útero y están presentes al nacer y, por lo tanto, no se someten a proliferación posnatal. Histológicamente, los hemangiomas congénitos no se tiñen con GLUT-1.
En cuanto a los hemangiomas congénitos, se describen tres tipos o formas de presentación: RICH, NICH y hemangioma congénito parcialmente involutivo (PICH). El RICH sufre involución espontánea durante el primer año de vida mientras que el NICH persiste de por vida.
La otra diferencia importante entre los HI y los congénitos es que los primeros no se deben a ninguna mutación conocida mientras que los segundos se deben a mutaciones en el espectro GNA (GNAQ / GNA11).
Hemangiomas infantiles sindrómicosAproximadamente el 30% de los pacientes con hemangiomas infantiles segmentarios faciales pueden desarrollar un síndrome con alteraciones en fosa posterior u otras lesiones estructurales del sistema nervioso central (SNC), hemangioma segmentario, anomalías arteriales, anomalías cardiacas, anomalías oculares y deformidades de la línea media del esternal (PHACES)4.
La evaluación debe incluir resonancia magnética cerebral, de cuello y tórax, evaluación oftalmológica, ecografía cardiaca y pruebas de función tiroidea.
Los hemangiomas sintomáticos de la vía aérea se asocian a una distribución segmentaria del hemangioma cutáneo en el área mandibular5.
Los hemangiomas segmentarios en la línea media inferior dorsal pueden estar asociados a anomalías congénitas renales, sacras, de la columna o genitourinarias, en lo que se conoce como síndrome LUMBAR o síndrome PELVIS6.
Tratamiento de los hemangiomasRespecto al tratamiento, los corticoides y la cirugía fueron durante medio siglo el tratamiento de elección para los HI complicados. En 2008 fueron descritos varios casos de involución de hemangiomas en pacientes tratados con propranolol oral, un betabloqueador no selectivo. El propranolol se convirtió en tratamiento de primera línea para el HI confirmándose su eficacia y seguridad en un ensayo clínico multicéntrico, randomizado y doble ciego que certificó que el propranolol era eficaz a una dosis de 3mg/kg/día, durante seis meses, para el tratamiento del HI, con mínimos efectos secundarios7.
Posteriormente, se han realizado numerosos estudios para identificar qué pacientes hay que tratar, cuándo hay que tratarlos y durante cuánto tiempo. En 2016, se publicó un consenso español sobre el manejo del hemangioma infantil que establece que deben tratarse los HI potencialmente mortales, los HI que comprometen la capacidad funcional, los HI ulcerados, con dolor o ausencia de respuesta a las medidas básicas de cuidado de la herida y aquellos con riesgo de desfiguración facial o del contorno corporal, que vayan a requerir cirugía reconstructiva futura. Se recomienda iniciar el tratamiento lo antes posible, durante un periodo de seis meses, realizando un seguimiento clínico y un ajuste de dosis por peso; aunque en algunos pacientes debe prolongarse más allá de los 12 meses8.
El tratamiento con láser o fuentes de luz puede reducir la coloración superficial de las telangiectasias residuales. También es útil, combinado con el propranolol, para el tratamiento de los hemangiomas ulcerados, haciendo desaparecer el dolor y favoreciendo el cierre de la úlcera ya desde la primera sesión9.
El manejo de los hemangiomas congénitos es conservador durante los primeros años (salvo en los casos de afectación hepática con insuficiencia cardiaca10, pero posteriormente, pasa a ser esencialmente quirúrgico, en las secuelas, tras la involución, o en aquellas lesiones que comprometan la funcionalidad o que presenten alteración cosmética significativa11,12.
Los HC no responden al propranolol ni a otros tratamientos farmacológicos conocidos hasta el momento actual, pero las mutaciones descubiertas pueden ser el primer paso para el tratamiento de aquellos HC que no involucionan, con inhibidores específicos13.
Otros tumores vascularesEl hemangioendotelioma kaposiforme (HEK) y el angioma en penacho son dos tumores vasculares estrechamente relacionados desde el punto de vista histopatológico y clínico, con un característico infiltrado linfático y el desarrollo, cuando su tamaño es grande, de trombocitopenia severa y coagulopatía (fenómeno de Kassabach-Merritt)14.
En 2010, se publicó el primer caso de tratamiento con éxito de un HEK grave con rapamicina15. Desde entonces, numerosas publicaciones han confirmado esta terapia, con mejoría clínica, recuperación del nivel de plaquetas, corrección de la coagulopatía y mínimos efectos secundarios por lo que ya se ha convertido en la primera línea de tratamiento16–19.
Malformaciones vascularesLas malformaciones vasculares son anomalías estructurales del aparato circulatorio y no tumores. Estas lesiones son, casi siempre, el resultado del desarrollo anómalo de la embriogénesis debido a una mutación y están presentes al nacimiento, aunque no siempre sean visibles. Estas anomalías evolucionan de diferentes formas y excepcionalmente involucionan. Básicamente se clasifican según el tipo de vaso afecto en:
- •
Malformaciones-arteriovenosas (MAV)
- •
Malformaciones-venosas (MV)
- •
Malformaciones-linfáticas (ML)
- •
Malformaciones-capilares (MC)
Las MAV son anomalías estructurales de alto flujo que forman comunicaciones anómalas entre el sistema arterial y el venoso. En neonatos pueden aparecer como una mancha rosada débil y pueden no ser fácilmente diferenciados de una MC. Las lesiones en tejidos más profundos pueden pasar desapercibidas durante años. Su ritmo de progresión depende de muchos factores entre los que se encuentra el tipo de mutación que las provoca (MAP2K1, KRAS, RASA1, BRAF, PTEN, entre otras) y va desde la estabilidad inicial a la insuficiencia cardiaca o el sangrado crítico final.
Las MAV que afectan las extremidades pueden presentar hipertrofia de hueso y partes blandas del miembro afecto20.
Malformaciones venosasLas MV son las anomalías del desarrollo de las venas que provocan casi siempre las mutaciones en TEK-TIE2 y más raramente otros genes como PIK3CA, glomulina o MAP3K321.
Las MV pueden clasificarse en:
- -
MV extratruncular o espongiforme: estas lesiones se pueden encontrar en la mayoría de los tejidos generalmente piel y músculo, en forma de lagos venosos conectados por microvénulas con el sistema venoso profundo22.
- -
MV troncular o flebectática: estas lesiones se presentan como dilataciones segmentarias o con presencia de venas aberrantes aneurismáticas (vena marginal del muslo o la vena ciática embrionaria persistente)23.
Las MV se asocian a coagulopatía intravascular localizada producida por la alteración del flujo sanguíneo a su través dando lugar a tromboflebitis, elevación del dímero D, formación de flebolitos y dolor incapacitante crónico24. Además, son siempre de crecimiento progresivo25, en parte inducido por la coagulopatía local26-28.
Malformaciones linfáticasLa nomenclatura de las ML sigue siendo confusa, y términos como linfangioma, linfangiectasia o displasia linfática están ya proscritos para describir las diversas entidades relacionadas21. Actualmente, las ML se clasifican según su presentación clínica, morfología y extensión en: malformación linfática simple, malformación linfática generalizada (con afectación ósea y visceral), trastornos de los grandes conductos linfáticos (quilotórax, ascitis quilosa, quilopericardio), linfangiomatosis kaposiforme, enfermedad de Gorham y linfedemas28,29.
La práctica mayoría están ocasionadas por mutaciones en PIK3CA, y un pequeño grupo, por mutaciones en KRAS y HRAS30.
Malformaciones capilaresLas MC son el tipo más común de AV y aparecen como una dilatación congénita de los vasos sanguíneos dérmicos superficiales que están presentes al nacer y crecen proporcionalmente al tamaño del niño. Permanecen de por vida y no tienen tendencia a la involución. La progresión clínica de las MC es variable y depende de la localización anatómica de la lesión.
Las MC pueden encontrarse en asociación con otras malformaciones vasculares y en localizaciones específicas se consideran una marca heraldo:
- -
MC fronto-orbitaria, malformación vascular meníngea y glaucoma: síndrome de Sturge Weber.
- -
MC en labio superior y narinas, megalencefalia y polimicrogiria: síndrome macrocefalia-malformación capilar.
- -
MC en el labio inferior, malformación linfática y venosa intraoral y cervical: síndrome capillary malformation of the lower lip, lymphatic malformation predominant on the face and neck, asymmetry, and partial/generalized overgrowth (CLAPO).
- -
MC en la línea media dorsal y malformación arteriovenosa medular: síndrome de Cobb.
- -
MC en la punta de los dedos y malformación linfático-venosa de la extremidad superior: síndrome FINCA (finger and nails capilar anomaly).
- -
MC en la cara lateral de tórax o muslos y malformación linfático-venosa con sobrecrecimiento: síndrome PROS (PIK3CA related overgrowth syndrome).
El tratamiento tradicional de las AV consistía en presoterapia, láser, embolización y tratamiento quirúrgico radical o paliativo. Estos tratamientos conseguían controlar parcialmente los síntomas, sin evitar la progresión, y en muchas ocasiones, sin poder mejorar la calidad de vida de los pacientes31.
La auténtica revelación en el manejo de las malformaciones vasculares en los últimos 10 años viene representada por las terapias dirigidas hacia la inhibición de las mutaciones presentes en cada subtipo de tumor o malformación32. El sirolimús o rapamicina es un inhibidor de la proteína mTOR (mamalian target of Rapamicyn) que, antes de su utilización en las AV, se empleaba fundamentalmente para evitar el rechazo de órganos en pacientes trasplantados renales. Además de ejercer como inmunomodulador, sirolimús posee un efecto antiangiogénico y antilinfoproliferativo que lo convierte en una herramienta más que adecuada para el tratamiento de las AV sintomáticas33. Inicialmente, fue probado en calidad de uso compasivo en anomalías refractarias a otros tratamientos, pero sus buenos resultados, especialmente en el campo de las malformaciones linfáticas, ha ampliado los objetivos y, actualmente, se utiliza, incluso como primera línea en multitud de patologías. En las ML simples, el sirolimús reduce el tamaño de la malformación, elimina la linforrea, disminuye el número de vesículas y reduce el número de episodios de sangrado intralesional e infecciones. En las ML complejas, el sirolimús estabiliza la osteólisis frenando su progresión y disminuye el débito de derrame pericárdico o pleural y de ascitis. Además, como ya se ha visto en el HEK, el sirolimús es capaz de corregir la coagulopatía. Aunque la vía habitual de administración es oral, a una dosis de 0,8mg/m2/12 h, también se ha demostrado mejoría en las ML microquísticas superficiales tras la administración de sirolimús tópico34.
Se ha confirmado que es un tratamiento eficaz y seguro, con mínimos efectos secundarios e inocuo, incluso en neonatos. Pero las preguntas sobre cuál es la dosis recomendada, qué niveles en sangre deben alcanzarse y durante cuánto tiempo se debe mantener el tratamiento aún continúan sin respuesta. La experiencia actual revela que el tratamiento con sirolimús debe ser individualizado y que el objetivo de dosis y niveles debe adaptarse al fenotipo y la sintomatología de cada paciente35.
Para reducir el dolor ante la presencia de trombos o flebolitos en malformaciones venosas se puede utilizar aspirina a 50 mg/día, aunque su eficacia no está bien descrita36. Si no hay respuesta, la administración de heparinas de bajo peso molecular, por vía subcutánea, es eficaz desde la primera dosis, recomendando su mantenimiento al menos durante dos semanas37. Actualmente se están utilizando cada vez más los anticoagulantes orales anti-Xa como rivaroxabán, aprobada su administración en niños, o anti-IIa como dabigatrán pudiendo mantenerse durante un tiempo en los casos más severos, y cuya combinación con rapamicina parece ser eficaz en la disminución del tamaño de estas lesiones38–40.
El descubrimiento de mutaciones en PIK3CA en AV ha hecho que los inhibidores de PIK3CA, desarrollados para el tratamiento del cáncer, hayan comenzado a utilizarse en estos pacientes con resultados prometedores.
Alpelisib es un inhibidor oral específico del phospatidil inositol 3 kinasa catalitic alfa (PI3KCA) de clase I que pertenece a la clase de compuestos 2-aminotiazol. En ensayos bioquímicos, alpelisib inhibe potentemente la subunidad p110α de la enzima phosphatidil 3 kinasa (PI3K) con un perfil de seguridad favorable. Alpelisib estaba solo aprobado en combinación con fulvestrant para el tratamiento de cáncer de mama avanzado o metastásico con mutación en PIK3CA receptor hormonal positivo (HR+), HER2 (human epidermal growth factor receptor 2), pero recientemente, la Administración de Alimentos y Medicamentos de EE. UU. (FDA) ha aprobado su uso en pacientes con AV congénitas que porten esta mutación. Canaud describió en 2018 un modelo de ratón posnatal de AV con sobrecrecimiento que recapitula parcialmente la enfermedad humana41,42. El modelo demostró la eficacia de alpelisib para prevenir y mejorar la disfunción orgánica. La publicación también describió cómo se utilizó el alpelisib en un programa de uso compasivo para tratar a 19 pacientes (adultos y pediátricos) con PROS, con mejora de los síntomas de la enfermedad en todos los pacientes tratados. Los tumores vasculares previamente intratables se hicieron más pequeños, se mejoró la insuficiencia cardiaca congestiva, se redujo la hemihipertrofia y se atenuó la escoliosis. El tratamiento no se asoció con efectos secundarios sustanciales. Esta serie de casos proporcionó la primera evidencia directa que respalda la inhibición de PIK3CA como una opción terapéutica prometedora en pacientes con PROS.
Actualmente, el tratamiento con alpelisib para pacientes con PROS se realiza bajo un programa de acceso al uso compasivo y en el contexto de cuatro ensayos clínicos. Teniendo en cuenta el número significativo de pacientes que están siendo tratados ya con alpelisib en esta indicación poco frecuente, se está trabajando en la revisión retrospectiva que evaluará los resultados clínicos y funcionales, así como la seguridad de dicho tratamiento.
Por ahora, los datos indican que un 75% de los pacientes responden al tratamiento, que un 25% muestran una reducción muy significativa de masas lipomatosas y que ningún paciente tratado ha abandonado el tratamiento debido a un efecto adverso grave42.
Como hemos apuntado previamente también se han encontrado en pacientes con malformaciones arteriovenosas y linfáticas mutaciones en KRAS, NRAS, BRAF y MAP2K1 que podrían derivar en futuros tratamientos dirigidos. Los inhibidores MEK1 que ya se utilizan en cáncer y neurofibromatosis tipo 1 pueden ser una opción de uso compasivo para estos pacientes que actualmente tienen pocas alternativas no invasivas. Trametinib y más recientemente selumetinib, son ya alternativas reales en pacientes con malformaciones de alto flujo, osteolisis y linfangiomatosis kaposiforme provocadas por mutaciones en KRAS43.
Nuevas moléculas e indicaciones están siendo exploradas, con destino a reducir la agresividad de tratamientos previos y de dar esperanza y calidad de vida a los pacientes sin respuesta a los tratamientos convencionales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

