







Introduction
Systemic lupus erythematosus (SLE) is a complex, multisystem autoimmune disease which results from the interplay of environmental, hormonal and genetic factors. In children, the presentation, clinical course and immunological findings differ slightly from adults with SLE 1.
In the last decade the outcome of SLE patients have improved remarkably, but even though many diagnostic and treatment options are similar for adults and children there are special issues that need to be considered in children and adolescents with SLE. For example, pSLE tends to be more severe and have higher impact on school adjustment and psychosocial aspects related, among others, to physical appearance and growth retardation 2.
Epidemiology
Pediatric SLE (pSLE) represents approximately 15-20 % of all SLE patients 1,3-5. It is more common in females than in males, with a female to male ratio varying from 2.3:1 to 9:1, depending on the study 6-11.
The incidence of the disease varies according to different ethnic groups. In Caucasian females the incidence of SLE with onset before age of 19 years is between 6 and 18.9 cases per 100,000, while it is 20-30 per 100,000 in African American females and 16-36.7 per 100,000 in Puerto Rican females 12. The diagnosis of pSLE is rare before the age of 10, and the average age at presentation is 12.1 years 6-11.
Disease damage and mortality in pSLE have been linked to different risk factors which include young age at diagnosis, male sex and non-white ethnicity (African American, Asian, and Hispanic) 5,13,14. African Americans tend to have a greater prevalence and a more severe renal and neuropsychiatric disease (NPSLE) 15. However, the association between any of these risk factors and a poor prognosis remains controversial 14,16.
The survival rate for pSLE has improved dramatically over the past 50 years, with a 5-year survival rate increasing from 50 % in 1955 to more than 90 % in 2004 8.
General clinical manifestations
Clinical characteristics and organ involvement vary depending on age of onset, gender and race. In general children with SLE tend to have a more severe disease at onset with higher rates of organ involvement and a more aggressive clinical course than adult-onset SLE patients 2,17.
At onset, 40-90 % of children will present with constitutional symptoms (fever, fatigue or weight loss), 20-82 % will have renal involvement, 20-74 % musculoskeletal symptoms, 22-74 % malar rash, 15-45 % lymphadenopathies and 15-74 % visceromegaly 4,7,9,18.
Cutaneous manifestations
The skin is commonly involved in pSLE. Different cutaneous manifestations have been reported in children during the course of their disease including: malar rash (22-74 %) which is a hallmark of SLE, oral ulcers (26-48 %), vasculitic rash (10-52 %), photosensitivity (16-50 %), alopecia (7-48 %), discoid lesions (5-19 %) and Raynaud's phenomenon (10-20 %) 4,7,9,18,19.
Musculoskeletal manifestations
Arthritis occurs in more than 3/4 of pediatric patients with SLE 2. It can be variable, but usually presents as a symmetric non-erosive, very painful polyarthritis involving both, large and small joints and is rarely associated with radiographic changes.
In general, SLE arthritis responds well to conventional therapy. Indeed, arthritis can be the only presenting manifestation of SLE and some patients who initially meet the American College of Rheumatology (ACR) criteria for juvenile arthritis, subsequently fulfilled clinical and serological criteria for the classification of SLE 2.
Myalgia is seen in 20-30 % of patients, however true myositis is less frequent. Musculoskeletal manifestations could be also seen as a side effect of the different drugs used. Treatment-induced musculoskeletal complications include avascular necrosis, osteoporosis and growth failure.
Hematological disorders
Thirty nine percent of children with SLE will develop hematological abnormalities, one of the ACR diagnostic criteria for SLE, during the course of the disease 20,21.
Autoimmune thrombocytopenia is the initial manifestation in up to 15 % of the pediatric cases although it can precede the onset of SLE by several years 1,3-5,21,22. It has been suggested that 20 % to 30 % of children with autoimmune idiopathic thrombocytopenic purpura who are ANA positive will evolve into SLE 21. Leukopenia is observed in 27-52 % of pediatric cases primarily due to a decrease in lymphocyte number. Granulocytopenia is also common 3.
Coagulation abnormalities are frequently present in pSLE. The Coomb's test is positive in approximately 30-40 % of patients, however less than 10 % will develop overt hemolysis 6. Antiphospholipid antibodies (aPL) are present in 75 % of pSLE patients 23. Pediatric patients with SLE and aPL, specifically lupus anticoagulant (LAC), are at high risk of developing thromboembolic events(TE) and the incidence of TE in LAC positive patients is 54 % 24. Therefore, lifelong anticoagulation must be considered after an initial thromboembolic event.
Cardiac manifestations
The spectrum of cardiac disease in pSLE mirrors that in adults with SLE. It encompasses 4 major types of manifestations: pericarditis (the most common form of cardiac involvement), myocarditis, valvular disease, and coronary artery disease due to either coronary arteritis or atherosclerosis 25. A few studies have reported silent cardiac abnormalities in children with SLE as a common finding 25,26. In fact, myocardial ischemia has been described in 16 % of asymptomatic children 26.
Cardiac involvement in pSLE patients is now being recognized as a major cause of morbidity and mortality in this population. Children with SLE have markedly higher rates of coronary heart disease than controls, and these increased rates are partly explained by an increase in the conventional cardiovascular risk factors 27. Identified risk factors for premature atherosclerosis in pSLE include: dyslipidemia, high levels of homocystein, presence of aPL, LAC, hypertension, hyperinsulinemia, nephritic range proteinuria, upregulated CD40-CD40 ligand interactions and steroid-induced obesity 27. Currently, the multicenter Atherosclerosis Prevention in Pediatric Lupus Erythematosus (APPLE) study group is assessing the role of statins for the prevention of atherosclerosis in the pediatric population with SLE.
Neuropsychiatric manifestations
Neuropsychiatric systemic lupus erythematosus (NPSLE), occuring in 20-45 % of children and adolescents, is the third most common cause for mortality in pSLE 2,28,29. Unlike other manifestations of the disease, central nervous system (CNS) involvement occurs within the first year of disease in approximately 75 % to 80 % of patients 2. NPSLE involvement can range from global cerebral dysfunction with paralysis and seizures, to mild or focal symptoms such as headache or memory loss 2. The presence of aPL is often linked to thrombosis and CNS infarction 30.
The diagnosis of NPSLE continues to be difficult since there is a lack of specific serologic studies for its diagnosis and monitoring 2. Although neuroimaging is a clinically useful tool, CSF analysis, EEG, CT scan, and even MRI may be normal in these patients 2. On the other hand, functional imaging may be abnormal in otherwise asymptomatic lupus individuals 28 which makes its interpretation difficult. Multiple imaging modalities have been studied to determine if there is a link between the clinical status and CNS imaging abnormalities, but to date there is no consensus 30.
Pulmonary involvement
The lung involved in 5-77 % of pSLE patients 29,31,32 and pulmonary manifestations vary from sub-clinical abnormalities to life-threatening disorders. The clinical spectrum includes pleuritis (the most common), pneumonitis, pneumonia, pneumothorax, diffuse interstitial disease, pulmonary hypertension and pulmonary hemorrhage, a relatively uncommon and potentially lethal complication. In the majority of children, pulmonary symptoms are present at some point during their disease course. Asymptomatic or subclinical lung involvement in pSLE may be more prevalent than realized. Pulmonary function abnormalities were found in up to 40 % of pSLE patients with no evidence of clinical symptoms or radiographic changes 31. The most frequent pattern observed was lung restrictive disease 28. Although pulmonary function tests do not correlate well with pulmonary symptoms, they provide objective quantification of the type and severity of the functional lesion observed 32.
Renal involvement
Not only does renal involvement represent the first clinical manifestation of the disease in 60-80 % of children with SLE 7,33, but it also determines the course of the disease and the outcome of patients. About 80 % of children and adolescents who develop renal abnormalities generally do so in the first year after diagnosis 7,33,34. Pathological findings can not be predicted from the clinical manifestations and therefore a renal biopsy is required for precise to establish diagnosis and subsequently planning effective therapy 33. In 1982, the World Health Organization (WHO) classified lupus nephritis (LN) based on histologic features into 6 categories 20. WHO class IV is the most common form of pSLE nephritis and is most commonly associated with the development of end stage renal disease or death.
Renal flares are common throughout the disease course of LN and can frequently be detected by increasing proteinuria. The presence of hypertension and peripheral edema are usually associated with WHO class III or IV LN 7. The prognosis of children with LN depends primarily on the severity of the histopathological lesions according to the WHO classification. Although in most centers the treatment is determined by the WHO class on biopsy, long-term renal outcome is still unpredictable. Other biopsy indices have been developed to evaluate LN at diagnosis and to predict outcome, including the National Institutes of Health (NIH) classification 35 and more recently an index focused on tubulointerstitial compartment changes in addition to features already included in the activity index and the chronicity indexes 36.
The prognosis of pediatric LN has improved dramatically in the past decade. The current 5-year survival rate for children with LN ranges between 78 % and 92 % 7,37 and the 5-year kidney survival from time of diagnosis varying from 44 to 93 % 7,35.
Diagnosis
The heterogeneous nature of lupus can result in a diagnostic challenge for physicians. Since there is not a single symptom or finding that in itself is sufficient for making the diagnosis of SLE, the ACR has developed different criteria that can be useful as general clinical guidelines for the initial assessment of patients with suspected SLE. The guidelines, created in 1982 and updated in 1997 (table 1), combine 11 criteria (clinical and laboratory) and a diagnosis can be made when four or more of these criteria are present 38.

It may be challenging to distinguish active inflammation from symptoms due to cumulative organ damage or medication side effects. A thorough history and physical examination, including all major systems, must be undertaken at each clinic visit (table 2). An assessment of disease activity is crucial for undertaking most treatment decisions. Several activity indices have been validated and are depicted in table 3.


Pathogenesis
Although the pathogenesis of SLE remains poorly understood, susceptibility involves a combination of environmental, hormonal and genetic factors. One of the environmental factors that has been involved in lupus is ultraviolet light, which triggers a photosensitive skin rash that may be followed by a generalized disease flare 39. There is also growing evidence for infections, such as Epstein-Barr virus (EBV) as an initial trigger for lupus-specific autoimmune responses. A higher incidence of EBV infection, higher titers of anti-EBV proteins and an abnormally elevated EBV load have been reported in children and adults with SLE when compared to matched healthy individuals 40,41.
Genetic factors
The genetic component of the disease is strongly established through epidemiology data, strong familial aggregation of SLE, and the known disease concordance rate in twins. Siblings of SLE patients have an increased relative risk of developing the disease when compared to the general population 42 and monozygotic twins have increased concordance (> 20 %) compared with dizygotic twins and other full siblings (2-5 %) 43,44. Through association and genetic linkage studies, over 60 loci 45-47, including alleles from the HLA region, region Fcg receptors and complement components have been implicated in the immunopathogenesis of SLE 45,48. Homozygous deficiency of any of the early components of the classical pathway (C1q, C1r, C1s, C4 and C2) have been linked with an increased predisposition to the development of SLE 49. Patients deficient in one of the C1-complex or C4 molecule exhibit the strongest prevalence (> 80 %) and the most severe disease, while strength of association and disease severity decrease in C2-deficient patients 49.
Immune alterations
It is recognized that SLE can arise through many molecular routes and requires contributions by T cells, B cells, dendritic cells, and nonlymphoid cells at sites of tissue injury. The most commonly recognized immune abnormalities are: the ability to produce pathogenic autoantibodies; lack of T-and B-lymphocyte regulation; and defective clearance of autoantigens and immune complexes by the immune system 39.
T cells
Although numerous abnormalities in T cell functions have been described in SLE, none are common to all patients. There is evidence of T-cell lymphopenia. Many studies report reduction of CD8+ T cells while others report decrease in CD4+ T cells; functional defects, such as decreased cytotoxic activity of CD8+ cells 50, signaling defects and diminished ability to reduce autoantibody production by B cells; sustained activation and abnormal cytokine production 39. T cells of SLE patients exhibit aberrant responses to stimuli with increased calcium influx and decreased production of interferon-a and IL-2 51. SLE T cells display markers of activation such as increased numbers of DR+ antigens 52 and are able to facilitate the production of immunoglobulins by B cells 53. SLE T cells seem to use different mechanisms of survival upon co-stimulation than normal T cells. Indeed, microarray profiling studies have recently shown that activated T cells from SLE patients resist anergy and apoptosis by upregulating cyclooxygenase-2 (COX-2) expression, which in turn increases c-FLIP (cellular homolog of viral FLICE inhibitory protein) and attenuates FAS signaling. Only certain COX-2 inhibitors, however, seem able to induce autoreactive T cell apoptosis and suppress the production of pathogenic autoantibodies to DNA in lupus-prone mice 54.
B cells and autoantibodies
B cells play a major role in the pathogenesis of SLE as they are responsible for the hypergammaglobulinemia and the production of antibodies against nuclear and cell surface antigens, one of the most prevalent immunological abnormalities in SLE. The development of some autoantibodies, such as anti-dsDNA antibodies, is closely linked to disease onset 51 while others, for instance antiphospholipid and anti-Ro antibodies, can be detected months to years preceding the development of SLE 55.
Patients with pSLE have profound B cell lymphopenia, involving both naïve and memory B cells, whereas oligoclonal plasma cell precursors are 3-fold expanded 56.
Antibody gene expression studies from single B cells from healthy individuals showed that large numbers of developing B cells in the bone marrow and recent emigrants in the blood express self-reactive antibodies. The majority of self-reactive B cells, however, are removed from the healthy mature blood naïve B cell pool at two discrete early checkpoints of their development 57. These checkpoints are defective in patients with SLE. A 25-50 % of the mature naïve B cells in SLE patients produce self-reactive antibodies even before they participate in immune responses as compared with 5-20 % in controls 58.
Dendritic cells
Individuals with SLE display major alterations in DC homeostasis. There is evidence that unabated IFN-α production differentiates CD14+ blood monocytes from SLE patients into mature dendritic cells able to capture dying cells and present their antigens to autoreactive T and B cells, leading to a break in tolerance 59,60.
Although only a fraction of patients with active disease show circulating IFN-α, microarray analyses demonstrate an IFN signature in blood mononuclear cells 60. These studies also demonstrated that high doses of glucocorticoids, broad inhibitors of immune cell function 61 extinguish the IFN signature. Preliminary studies by Palucka et al show that they induce the apoptosis of interferon-producing cells or dendritic cells (Palucka et al unpublished). Glucocorticoids may therefore, act by blocking IFN-α production.
Apoptosis
A common feature of SLE autoantigens is that they are components of the surface blebs 62 and they come under immune surveillance when they arise to the surface of apoptotic cells. Furthermore, there is increasing evidence that apoptotic material is normally taken up by immature dendritic cells and cross-presented to induce T cell tolerance 63. Deficiency in apoptotic cell removal may provide dendritic cells with an excessive load of nuclear antigens and consequently develop overt SLE.
Several other factors that are linked to the pathogenesis of SLE can influence apoptosis such as estrogens, UV light 64, infections 65 and autoantibodies themselves 66.
Treatment
Treatment of SLE depends on the clinical manifestations and the presence/absence of major organ involvement (table 4). Corticosteroids are a major cause of morbidity and mortality in pSLE but they continue to be a mainstay of treatment due to their dramatic and rapid impact on lupus flares. Their effectiveness in treating SLE has been recognized since the 1950s. Intravenous (IV) pulse methylprednisolone (MEP) can be successfully used to treat major organ involvement and/or life-threatening manifestations of SLE. Antimalarials are effective for milder manifestations and improve bone-mineral density and dyslipoproteinemia 67. Cyclophosphamide (CYC) remains the first-line treatment for major organ involvement. It has been shown to reduce morbidity and improve mortality in lupus patients. Over 20 years ago a National Institute of Health (NIH) study 68 showed that monthly IV pulses of CYC were as effective, but less toxic, than daily oral CYC. Since then the gold standard immunosuppressive treatment of LN has been monthly IV pulse CYC for 6-7 months, in combination with high-dose glucocorticoids, followed by a 2-year maintenance phase (CYC for 2-3 months). All patients receiving CYC and high-dose glucocorticoids should also receive prophylaxis with low-dose trimethoprim-sulfametoxazole inorder to prevent the most common opportunistic infection, Pneumocystis jiroveci pneumonia.
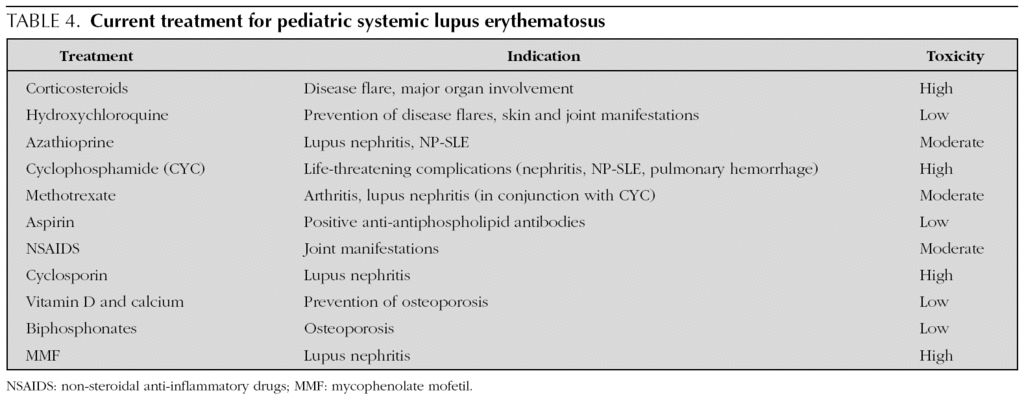
Treatment of SLE includes not only pharmacological therapies, but also patient education, such as protection from ultraviolet light, management and prevention of infections, cardiovascular risk factors, and treatment of complications including osteoporosis.
Although the prognosis for pSLE has improved over the last few years, pSLE remains a very challenging disease, especially in children with partial response to treatment or treatment resistant who are at a high risk for serious complications. With better understanding of SLE pathogenesis, novel therapies are emerging which will hopefully translate into safer and more efficient treatments for children with SLE (table 5).

